


Vol. XXXI Issue 1
Article 1
ARTÍCULOS ORIGINALES
Enfermedad de Creutzfeldt-Jakob de origen genético: serie de casos en la Patagonia Argentina
Creutzfeldt-Jakob syndrome of genetic origin: series of cases in the Argentinian Patagonia
Exeni Díaz G.1, Costa M.1, Salman J.2, Ávila S.1,3
1 Servicio de Genética, Hospital
Provincial Neuquén, Argentina.
2 Centro Médico Integral, Neuquén,
Argentina.
3 Facultad de Ciencias Médicas,
Universidad Nacional del Comahue,
Argentina.
Corresponding author:
Mailén Costa
mailencosta89@gmail.com ORCID 0000-0002-1690-8879
DOI: 10.35407/bag.2020.31.01.01
Received: 11/10/2018
Revised version received:
07/01/2019 - 03/11/2020
Accepted: 04/05/2020
RESUMEN
La enfermedad de Creutzfeldt-Jakob (CJD) es un desorden neurodegenerativo poco frecuente con una incidencia estimada en 1 por cada 1.000.000 por año, típicamente caracterizado por demencia rápidamente progresiva, ataxia, mioclonus y cambios de comportamiento. Las enfermedades genéticas priónicas se desarrollan debido a mutaciones en el gen de proteína priónica PRNP. Entre el 10 y el 15% corresponden a formas familiares que se transmiten con patrón autosómico dominante con alta penetrancia. La mutación más frecuente a nivel mundial es la E200K (glutamato por lisina). Se reportan cuatro familias con CJD que fueron atendidas en el Hospital Provincial Neuquén en el año 2018. Tres de los cuatro casos índice tenían historia familiar de trastornos neurológicos y psiquiátricos pero estos datos no fueron jerarquizados en la evaluación inicial del caso. Se consideró la consulta genética por la edad temprana de presentación de la enfermedad. En todos los casos la consulta fue solicitada por el neurólogo. Los síntomas iniciales que presentaron los pacientes fueron insomnio pertinaz y depresión con pobre respuesta a la medicación psiquiátrica habitual. En todos los casos la progresión de la enfermedad fue rápida con desórdenes visuales, mioclonías, ataxia, demencia y pérdida del lenguaje. El análisis de los pedigrees permitió identificar ciento cuarenta personas que potencialmente podrían portar el gen y desarrollar la enfermedad en algún momento de la vida adulta. En todos los casos se identificó la mutación E200K. En la región existe una frecuencia aumentada de CJD. Debe ser investigada en los pacientes con síntomas neuropsiquiátricos e historia familiar sospechosa. Los estudios genéticos confirman el diagnóstico en los pacientes y permite identificarlos en individuos en etapa presintomática. Esto plantea un desafío para el asesoramiento genético familiar y para evitar la transmisión iatrogénica del trastorno.
Palabras clave: Enfermedad de Creuztfeldt-Jakob familiar; Gen PRNP; E200K; CJD en Patagonia.
ABSTRACT
Creutzfeldt-Jakob disease (CJD) is an uncommon neurodegenerative disorder with an incidence of 1 per 1,000,000 in humans per year, typically characterized by rapidly progressive dementia, ataxia, myoclonus and behavioral changes. Genetic prion diseases, which develop due to a mutation in the prion protein gene (PRNP), account for an estimated 10 to 15% of all CJD cases. Familial CJD is transmitted with an autosomal dominant inheritance pattern with high penetrance. Worldwide, the most common mutation is E200K (glutamate to lysine). We report four families with CJD assisted in Neuquén Hospital in 2018. Three of the four index cases had family history of neurological and psychiatric illness, though data was not taken into consideration at the moment of evaluation of the new cases. The most significant data recorded for a genetic consultation was when the problem had started, and it was required by a neurologist. The initial symptoms were persistent insomnia and depression with poor response to habitual psychiatric medication. Impoverishment is fast with visual disorder, myoclonias, ataxia, dementia and loss of language. Pedigree analysis allowed the identification of 144persons with the gene potential, who can develop the disease at any time in their adulthood. In all cases, mutation E200K was identified. There is a region of increased frequency of CJD. There must be suspicion on patients with neuropsychiatric symptoms and suspected family history(familiar background). Finding of the mutation confirms the diagnosis in patients and allows the identification on pre-symptomatic individuals. Challenge is posed on gene advice and to avoid iatrogenic disorder transmission.
Key words: Familial Creutzfeldt-Jakob Syndrome; PRNP gene; E200K; CJD in Patagonia.
INTRODUCCIÓN
La enfermedad de Creutzfeldt-Jakob (CJD) es un
trastorno neurodegenerativo causado por priones en
humanos. Se presenta como encefalopatía espongiforme
de distribución mundial con una prevalencia de 1 en
1.000.000 de habitantes (Prusiner y Hsiao, 1994; Prusiner,
2001). Las formas familiares (fCJD), que se transmiten
con patrón de herencia autosómico dominante con
alta penetrancia, dan cuenta de entre el 10 y el 15% de
las enfermedades priónicas (Mastrianni, 2003). En las
formas hereditarias la proteína priónica, codificada por
el gen mutado, es capaz de modificar el plegamiento
de otras proteínas normales; esta propiedad hace que
esta enfermedad pueda ser transmitida a personas no
emparentadas a través de fluidos corporales.
Los priones pueden colonizar diferentes tejidos, pero
el único sistema en el cual se ha podido demostrar daño
histopatológico, tanto en animales como en humanos,
es el sistema nervioso central.
Clínicamente CJD se caracteriza por el desarrollo de
una demencia rápidamente progresiva asociada con
ataxia mioclónica.
El único gen vinculado con la forma genética es PRNP.
Se han descrito más de 40 mutaciones germinales
en el gen PRNP pero la más frecuente es c.598G>Ap.
Glu200Lys (E200K).
Se describen clusters con alta prevalencia de fCJD
en Eslovaquia, Libia, Túnez, Chipre, Alemania, Sicilia,
Austria, Japón y Chile (Takada, 2017; Meiner et al., 1997;
Lee et al., 1999; Mitrova y Belay, 2002).
Lee et al (1999) estudió el origen de la mutación E200K
a partir de los estudios de ancestralidad de 62 familias
de 11 poblaciones diferentes. Concluyó que los clusters
de Libia, Túnez, Italia, Chile y España comparten un
haplotipo mayor; esto sugiere que la mutación podría
haberse originado de un mismo evento mutacional,
probablemente en la Península Ibérica, y a partir de allí,
podría haberse expandido hacia el Mediterráneo y los
países de América del Sur.
La sospecha de fCJD se basa en una combinación de
síntomas neuropsiquiátricos, historia familiar positiva y
mutación en el gen PRNP. Los estudios de neuroimágenes,
EEG y examen de LCR pueden ser orientativos y permiten
plantear diagnósticos diferenciales con otros trastornos
neurodegenerativos (CDC, 2015).
En el reporte del Centro de Referencia en Enfermedad de
Creutzfeldt-Jakob de Argentina se informó un aumento de
la frecuencia esperada de pacientes con fCJD en la región
patagónica en coincidencia con lo detectado en Chile
(Begue, 2011). Las acciones del Centro de Referencia no
incluyeron el asesoramiento genético para los familiares
de los casos índices identificados ni la participación de
los servicios de genética.
El objetivo de este trabajo es reportar cuatro familias
estudiadas durante el año 2018 en el Hospital Provincial
Neuquén.
REPORTE DE SERIE DE CASOS
Las cuatro familias fueron estudiadas por el Servicio de Genética del Hospital Provincial Neuquén durante el año 2018. En la Tabla 1 se resumen los principales hallazgos clínicos, electroencefalográficos y de neuroimágenes presentes en los casos índice.
Tabla 1. Síntomas y resultados de estudios complementarios al momento de la confirmación diagnóstica.
*NR no reportado.
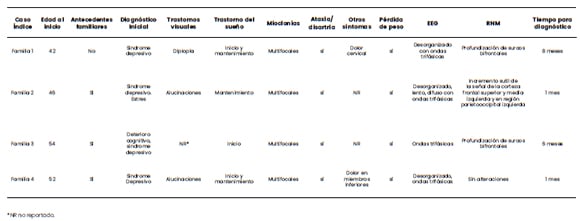
Familia 1
El caso índice es una mujer de 42 años, oriunda de
Neuquén, la mayor de una hermandad de dos. Su
padre y madre están sanos, los abuelos maternos
fallecieron en la octava década de vida con trastornos
cognitivos (Figura 1). En la rama materna los abuelos
proceden de Chile y en la rama paterna de Italia. El
cuadro se inicia con trastornos del sueño y ansiedad
que motivan su consulta con especialista en salud
mental y medicación con inhibidores de la recaptación
de serotonina sin respuesta terapéutica. Presenta
cefalea de intensidad progresivamente en aumento y
diplopía. Aproximadamente a los treinta días se agregan
mioclonías, ataxia y alteraciones del lenguaje. Se produce
marcada pérdida de peso. A los sesenta días del inicio del
cuadro la paciente se encuentra en mutismo aquinético.
Se realizaron dos estudios de RNM sin alteraciones
y EEG con trazado desorganizado y ondas trifásicas.
A los noventa días del inicio de los síntomas y luego de
descartar otras causas de encefalopatía se solicita la
evaluación de Genética.
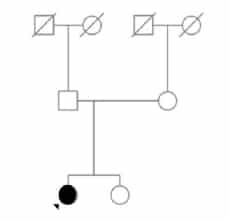
Figura 1. Familia 1
Familia 2
El caso índice es una mujer de 46 años oriunda de
Zapala, con antecedente de madre fallecida en la sexta
década de vida con probable CJD. La paciente tiene tres
hijos mayores de edad y seis tíos por vía materna, uno de
ellos fallecido con un cuadro de demencia rápidamente
progresiva. Los síntomas iniciales fueron insomnio
y ansiedad que se atribuyeron al estrés de su trabajo.
Realiza consulta con psicología, luego con psiquiatra y
ante la falta de respuesta a la medicación se solicita la
evaluación con Neurología. En algunos días se suman
trastornos visuales, mioclonías, ataxia y deterioro
cognitivo. En los dos meses siguientes pierde la marcha
y el lenguaje hasta configurar un cuadro de mutismo
aquinético. En ese momento se solicita la evaluación por
Genética. El análisis de la genealogía muestra al menos
30 familiares, muchos de ellos en edad fértil, que podrían
haber heredado la mutación (Figura 2).
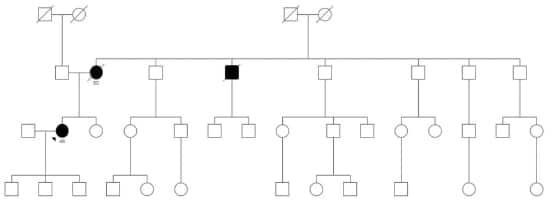
Figura 2. Familia 2
Familia 3
El caso índice es una mujer de 54 años oriunda de
Cutral Có que consulta para asesoramiento genético
por haber en su familia varios casos de afectados
por CJD, incluyendo su propia madre. La paciente
manifiesta querer conocer su riesgo de afección para
poder planificar su vida. Su madre falleció de CJD a los
sesenta y cuatro años al igual que una tía y tres tíos
segundos. Por su propia iniciativa ha conseguido, ocho
años después de realizados en el Centro de Referencia
en Enfermedad de Creutzfeldt-Jakob, los informes de los
estudios de uno de sus tíos fallecidos con la enfermedad;
la investigación de la mutación E200K está informada
como positiva. Refiere que los síntomas iniciales en su madre fueron insomnio y trastornos cognitivos con
crisis de excitación por lo cual se indica internación en
centro de psiquiatría donde permanece por treinta días.
Ante su empeoramiento se deriva a un centro de atención
clínica donde es evaluada por Neurología y se realiza el
diagnóstico de CJD probable. Ya en mutismo aquinético
se define tratamiento domiciliario y logra una sobrevida
de un año. La genealogía de esta familia es extensa y
revela al menos 84 personas que podrían ser portadoras
de la mutación (Figura 3). Los bisabuelos maternos de
la consultante eran de origen chileno y se radicaron en
Neuquén en el siglo XIX.

Figura 3. Familia 3
Familia 4
El caso índice es un hombre de 52 años, oriundo de
Neuquén, el mayor de cinco hermanos, todos ellos con
hijos y nietos. Su padre, de nacionalidad chilena, falleció
en la séptima década de vida con un cuadro de demencia
que evolucionó con óbito a los tres meses del inicio
(Figura 4). Desconocen otros antecedentes familiares.
Los síntomas del caso índice se inician con insomnio
y depresión. En pocos días se agregan mioclonías y
trastornos cognitivos. Evoluciona rápidamente con
el óbito a los tres meses del inicio de los síntomas. La
RNM muestra patrón característico de CJD y el EEG el
patrón de ondas trifásicas. La consulta con Genética fue
realizada al mismo tiempo que se solicitaba la evaluación
neurológica.
En las cuatro familias se pudo corroborar la
segregación de la mutación E200K.
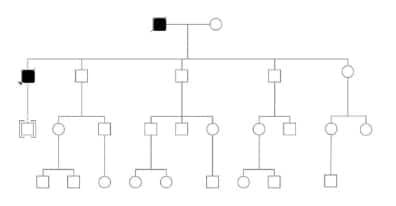
Figura 4. Familia 4
DISCUSIÓN
La enfermedad de Creutzfeldt-Jakob tiene una ocurrencia
mundial estimada en un caso por millón de habitantes.
Existen clusters descriptos con alta prevalencia debido
a la segregación de la mutación E200K por un efecto
fundador ya planteado por Lee et al (1999) a partir de
estudios de ancestralidad. Begue (2011) reporta que el
número de casos familiares de la Patagonia es alto y está
en relación con el cluster que se reconoce en Chile. Sin
embargo esta observación no contempló la intervención
de médicos genetistas en el diseño de protocolos de
estudio y tratamiento, ni acciones de difusión sobre
los equipos de salud para concientizar acerca de la
naturaleza de esta patología, de modo de acortar los
tiempos diagnósticos, asesorar a las familias de riesgo y
evitar la transmisión iatrogénica.
En las cuatro familias que describimos en este reporte
hay por lo menos un familiar que se refiere como oriundo
de Chile a finales del siglo XIX y comienzos del XX. El
intercambio poblacional entre ambos países es frecuente
en las provincias limítrofes de modo bidireccional:
las fronteras establecidas por los Estados no son las
que se establecen en las relaciones interpersonales. La
mutación encontrada en las cuatro familias corresponde
a la E200K que es la que se ha descripto con mayor
frecuencia en Chile.
Los datos reportados en la bibliografía son
controversiales en lo que respecta a la penetrancia
(Minikel, 2016) pero en tres de las cuatro familias que
presentamos se observó un adelantamiento de al menos
veinte años en la presentación inicial de los síntomas en
los casos índice con respecto a sus progenitores.
La evolución clínica de los individuos de nuestras
familias es muy rápida a partir de los primeros síntomas
que son fundamentalmente psiquiátricos con insomnio
pertinaz, depresión y trastornos visuales, sin respuesta
a la medicación habitual. Posteriormente se agrega
deterioro cognitivo y ataxia mioclónica.
CONCLUSIÓN
A pesar de haberse reportado el aumento de la
frecuencia de CJD en la región patagónica a expensas
de casos familiares, esta información no es conocida
ampliamente por los profesionales del equipo de salud.
Si bien es cierto se había reportado la existencia de casos
familiares en la Patagonia pero hasta la descripción de
estos casos no se habían implementado estrategias para
lograr una sospecha rápida, accesibilidad diagnóstica
y abordaje óptimo de los familiares en riesgo de portar
y transmitir la mutación. Esto se evidencia porque, a
pesar de ser referidos los antecedentes familiares, la
posibilidad de enfermedad genética fue considerada sólo
cuando se excluyeron otras causantes de encefalopatía
rápidamente progresiva. Siguiendo las pautas de la OMS
y del CDC ante la presencia de antecedentes familiares
de CJD y síntomas neuropsiquiátricos, se deben tener en
cuenta los estudios genéticos en el plan de estudios.
El diagnóstico de fCJD tiene implicancias individuales,
familiares y epidemiológicas.
Las implicancias individuales son similares a las
descriptas en otras enfermedades neurodegenerativas
de inicio en el adulto. Los pacientes que fueron testigos
del avance de la enfermedad en sus familiares suelen
reconocer en sí mismos los primeros síntomas aún antes
que los mismos se hagan evidentes para el entorno.
Desde el punto de vista familiar, cuando se recibe el
diagnóstico de CJD, aparece conmoción, confusión y
angustia. Los familiares deben aceptar rápidamente
la noticia de que su ser querido, saludable hace unas
semanas, padece una enfermedad terminal que no tiene
tratamiento. La naturaleza genética del trastorno agrega
la culpa y la angustia de quienes empiezan a entender
que tienen a priori una probabilidad del 50% de portar el
gen y una alta probabilidad de desarrollar la enfermedad
en algún momento de la vida. Es esencial la presencia
del médico genetista para el asesoramiento genético
pre y post test dado el impacto que puede tener tanto un
resultado positivo como negativo (Roberts y Uhlmann,
2013).
Desde el punto de vista epidemiológico, la
identificación de la propia población como un cluster de
CJD plantea un desafío a las autoridades y a los equipos
de salud.
A partir de los datos del presente reporte en el sistema
de salud de la Provincia de Neuquén se pone en marcha de
modo gratuito, el diagnóstico molecular de la mutación
E200K en los pacientes con sospecha clínica de fCJD.
Se define reforzar las estrategias para sensibilizar a los
equipos de salud para la detección de casos sospechosos
ante la existencia de antecedente familiar positivo y
cuadro clínico compatible, a los fines de realizar los test
genéticos dentro de los estudios iniciales. Este aspecto
es importante para evitar la demora diagnóstica y la
realización de estudios complementarios innecesarios.
La realización de las pruebas moleculares en el ámbito
de la salud pública garantiza no sólo la accesibilidad a
las mismas sino también la posibilidad de perfeccionar
la vigilancia epidemiológica de la enfermedad.
Se define también trabajar en una red de atención
interdisciplinaria para las personas en riesgo de portación
del gen; esta red incluirá el acceso al asesoramiento
genético pre- y post-test y el acompañamiento psicosocial.
BIBLIOGRAFÍA
1. Begue C. (2011) Creutzfeldt-Jakob disease surveillance in Argentina 1997-2008. Neuroepidemiology 37 (3-4): 193-202.
2. CDC (2015) Creutzfeldt-Jakob disease. https://www.cdc.gov/prions/cjd/index.html.
3. Kóvacs G. (2002) Mutations of the prion protein gene phenotypic spectrum. J. Neurol. 249: 1567-1582.
4. Lee H.S., Sambuughin N., Cervenakova L., Chapman J., Pocchiari M., Litvak S., Qi H.Y., Budka H., del Ser T., Furukawa H., Brown P., Gajdusek D.C., Long J.C., Korczyn A.D., Goldfarb L.G. (1999) Ancestral origins and wordwide distribution of the PNRP 200K mutation causing familial Creutzfeldt-Jakob disease. Am. J. Hum. Genet. 64 (4): 1063- 1070.
5. Mastrianni J.A. (2003) Genetic Prion Diseases. Gene Reviews[Internet], Seattle (WA): University of Washington, Seattle; 1993- 2018. https://www.ncbi.nlm.nih.gov/books/NBK1229/ (accesed October 2018).
6. Meiner A, Gabizon R., Prusiner S. (1997) Familial Creutzfeldt-Jakob disease. Codon 200 prion disease in Libyan Jews. Medicine (Baltimore) 76 (4): 227-237.
7. Minikel E.V. (2016) Quantifying prion disease penetrance using large populations controls cohorts. Sci. Transl. Med. 8 (322): 322-ra329.
8. Mitrova E., Belay G. (2002) mutation Creutzfeldt-Jakob disease with E200K in Slovaki: Characterization and development. Acta Virol. 46 (1): 31-39.
9. Prusiner S. (2001) Neurodegenerative diseases and prions. N. Engl. J. Med. 344: 1516.
10. Prusiner S., Hsiao K. (1994) Human prion diseases. Ann. Neurol. 35: 385-395.
11. Roberts S., Uhlmann W. (2013) Genetic susceptibility testing for neurodegenerative diseases: Ethical and practice issues neurodegenerative diseases. Prog. Neurobiol. 110: 89-101.
12. Rosenmann H, Kahana E., Korczyn A.D., Kahana I., Chapman J., Gabizon R. (1999) Preliminary evidence for anticipation in genetic E200K Creutzfeldt-Jakob disease. Neurology 54 (6): 1328-1329.
13. Takada L. (2017) Genetic prion disease: Experience of a rapidly progressive dementia center in the United State and review of the literature. Am. J. Med. Genet. Part B 174B: 36-69.