


Vol. XXXI Issue 1
Article 3
ARTÍCULOS ORIGINALES
Meta-análisis para evaluar eficiencia de selección genómica en cereales
Meta-analysis for evaluating the efficiency of genomic selection in cereals
Rueda Calderón M. A.1,2 , Balzarini M.1,2, Bruno C.1,2*
1 Estadística y Biometría. Facultad
de Ciencias Agropecuarias.
Universidad Nacional de Córdoba,
Córdoba, Argentina. Ing. Agr.
Félix Aldo Marrone 746. Ciudad
Universitaria, Córdoba, Argentina
2 Unidad de Fitopatología
y Modelización Agrícola
(UFyMA), Consejo Nacional de
Investigaciones Científicas y
Técnicas (CONICET), Córdoba,
Argentina.
Corresponding author:
Cecilia Bruno
cebruno@agro.unc.edu.ar
DOI: 10.35407/bag.2020.31.01.03
Received: 03/09/2020
Revised version received: 04/23/2020
Accepted: 04/28/2020
RESUMEN
La selección genómica (SG) es usada para predecir el mérito de un genotipo respecto a un carácter cuantitativo a partir de datos moleculares o genómicos. Estadísticamente, la SG requiere ajustar un modelo de regresión con múltiples variables predictoras asociadas a los estados de los marcadores moleculares (MM). El modelo se calibra en una población en la que hay datos fenotípicos y genómicos. La abundancia y la correlación de la información de los MM dificultan la estimación, y por ello existen distintas estrategias para el ajuste del modelo basadas en: mejor predictor lineal insesgado (BLUP), regresiones Bayesianas y aprendizaje automático. La correlación entre el fenotipo observado y el mérito genético predicho por el modelo ajustado, provee una medida de eficiencia (capacidad predictiva) de la SG. El objetivo de este trabajo fue realizar un meta-análisis de la eficiencia de la SG en cereales. Se realizó una revisión sistemática de estudios relacionados a SG y se llevó a cabo un meta-análisis, para obtener una medida global de la eficiencia de la SG en trigo y maíz, bajo diferentes escenarios (cantidad de MM y método estadístico usado para la SG). El metaanálisis indicó un coeficiente de correlación promedio de 0,61 entre los méritos genéticos predichos y los fenotipos observados. No se observaron diferencias significativas en la eficiencia de la SG realizada con modelos basados en BLUP (RR-BLUP y GBLUP), enfoque estadístico más comúnmente usado. El incremento de MM no cambia significativamente la eficiencia de la SG.
Palabras clave: Revisión sistemática; Modelos de efectos aleatorios; Forest plot; Capacidad predictiva.
ABSTRACT
Genomic selection (GS) is used to predict the merit of a genotype with respect to a quantitative trait from molecular or genomic data. Statistically, GS requires fitting a regression model with multiple predictors associated with the molecular markers (MM) states. The model is calibrated in a population with phenotypic and genomic data. The abundance and correlation of MM information make model estimation challenging. For that reason there are diverse strategies to adjust the model: based on best linear unbiased predictors (BLUP), Bayesian regressions and machine learning methods. The correlation between the observed phenotype and the predicted genetic merit by the fitted model provides a measure of the efficiency (predictive ability) of the GS. The objective of this work was to perform a metaanalysis on the efficiency of GS in cereals. A systematic review of related GS studies and a meta-analysis, in wheat and maize, was carried out to obtain a global measure of GS efficiency under different scenarios (MM quantity and statistical models used in GS). The meta-analysis indicated an average correlation coefficient of 0.61 between observed and predicted genetic merits. There were no significant differences in the efficiency of the GS based on BLUP (RR-BLUP and GBLUP), the most common statistical approach. The increase of MM data, make GS efficiency do not vary widely.
Key words: Systematic review; Random effects model; Forest plot; Predictive accuracy.
INTRODUCCIÓN
La selección genómica (SG) es una técnica con alto
potencial para acelerar la tasa de ganancia genética en
vegetales (Heffner et al., 2009). Valiéndose de modelos
estadísticos, permite relacionar vasta cantidad de
marcadores moleculares (MM) o información genómica
a un carácter fenotípico de interés, para predecir luego
el mérito genético de cada fenotipo. En SG se parte de
una población de entrenamiento o calibración, donde
no sólo el genotipo molecular es conocido sino también
el fenotipo, y se estiman modelos relacionales, que
aprendiendo desde dicha población son luego aplicados
a poblaciones de líneas donde no se conoce el fenotipo
pero si se desea predecir el mérito genético. Es a partir del
modelo estadístico estimado o ajustado que se realizan
predicciones para estimar el valor de cría o mérito
genético de cada individuo en la población de interés.
Así, es posible seleccionar individuos con características
promisorias para un determinado carácter, usando sólo
la información molecular que usualmente proviene del
genotipado con marcadores moleculares distribuidos en
todo el genoma de cada individuo (Hawkins y Yu, 2018).
La información del genotipo y del fenotipo en la población
de entrenamiento también es usada para estimar el
efecto de cada marcador sobre el carácter de interés.
Aun con gran cantidad de MM, los modelos estadísticos
usados en SG permiten ajustar el efecto de todos los
marcadores simultáneamente. Si los MM se encuentran
en desequilibrio de ligamiento con la mutación que afecta
el carácter, los marcadores serán capaces de capturar
una gran proporción de la varianza genética del carácter
de interés (Voss Fels et al., 2019) y el modelo estadístico
permitirá asociar los MM con el fenotipo (Hawkins y Yu,
2018). El modelo aplicado sobre la población de mejora
se usa para predecir el valor genético del individuo desde
la información molecular distribuida en todo el genoma
(Genome Estimated Breeding Value-GEBV) (Bhat et al.,
2016; Hawkins y Yu, 2018). La SG tiene la capacidad de
utilizar altas cantidades de MM asociados a cada loci,
incluso de efecto menor (Heffner et al., 2009) y así
capturar mayor variación genética.
La eficiencia de la SG en un contexto particular,
puede evaluarse a través de las correlaciones entre los
fenotipos observados y los méritos genéticos predichos o
valor de cría (GEBV). Existen una importante cantidad de
estudios primarios de SG en vegetales, y si bien es posible
su recopilación a través de la revisión sistemática de
literatura, no es común encontrar estudios secundarios
o meta-análisis (MA) de esos estudios primarios. La
revisión sistemática permite sintetizar la información
científica disponible, pero es el MA la técnica que permite
incrementar la potencialidad de las conclusiones de los
estudios primarios (Ferreira González et al., 2011).
La revisión sistemática involucra una serie de
acciones: i) formular la pregunta de investigación a
partir de la cual se realiza un constructo de búsqueda
para explorar plataformas digitales; ii) realizar la
búsqueda de manera exhaustiva y comprensiva de
estudios primarios en diferentes bases de datos; iii)
compactar la información obtenida de las diferentes
bases de datos a través de un gestor bibliográfico; iv)
establecer los criterios de exclusión e inclusión para
la selección de estudios primarios; v) determinar la
relevancia de los estudios identificados; y vi) extraer los
datos en el formato necesario para el MA que se quiera
realizar (Pai et al., 2004; Akobeng, 2005; Borenstein
et al., 2009; 2010; Sánchez Meca, 2010). El término
MA fue introducido por primera vez por Glass (1976),
para denotar la síntesis estadística de los resultados de
estudios primarios similares respecto al tema objeto de
estudio. El MA permite: i) sintetizar los resultados de
estudios primarios obtenidos de la revisión sistemática
para incrementar la potencia de las pruebas de hipótesis
de interés; ii) estimar el tamaño del efecto de interés o
efecto global; iii) evaluar heterogeneidad entre estudios;
y iv) en caso de que sea necesario, hacer análisis por
subgrupos o meta-regresiones para comprender el
impacto de factores y covariables sobre el tamaño del
efecto en cada estudio primario (Borenstein et al., 2009).
El MA permite estimar un efecto global o de síntesis de
los estudios primarios. En este trabajo, el efecto que se
midió a través del MA de estudios de SG es el tamaño
o magnitud de la correlación entre valores fenotípicos
observados y méritos genéticos predichos por los
modelos de SG en las poblaciones de mejora. El gráfico
Forest Plot, permite visualizar los resultados del MA a
través de intervalos de confianza para el valor esperado
del efecto de interés, tanto para cada estudio primario
como para el conjunto de éstos. La amplitud de estos
intervalos de confianza dependerá de la precisión con
que se reportan los resultados de cada estudio primario;
ésta es función del tamaño muestral y de la varianza
residual. El efecto global es estimado como una media
ponderada de los efectos reportados en los estudios
primarios, y esta ponderación depende de la precisión de
cada estudio. En escenarios de alta heterogeneidad entre
estudios respecto al tamaño del efecto objeto de estudio,
se realizan análisis por subgrupos de estudios primarios
relativamente homogéneos. En los estudios de SG en
vegetales, es frecuente que se presenten datos de otras
variables que podrían marcar diferencias o generar
heterogeneidad entre estudios, como por ejemplo,
la cantidad de genotipos evaluados, la cantidad de
marcadores moleculares empleados (Wu y Hu, 2012) o los
modelos estadísticos usados para la SG. Esta información
puede ser considerada para conformar subgrupos para el
MA y así comparar el impacto de los distintos niveles de
estos factores sobre la correlación objeto de estudio. El
objetivo de este trabajo fue realizar un MA de la eficiencia
de la SG en cereales y su dependencia con la cantidad de
MM y el modelo estadístico usado.
MATERIALES Y MÉTODOS
Recolección de la información a través de la revisión
sistemática
En la revisión sistemática se seleccionaron palabras
clave relacionadas a la pregunta de investigación, y de
este compendio de palabras se construyó el siguiente
constructo de búsqueda: (GS o “Genomic Selection”)
y (“Plant breeding”) y (crops) para obtener la mayor
cantidad posible de literatura referente a estudios de
SG en mejoramiento vegetal. La búsqueda de estudios
primarios fue llevada a cabo en múltiples bases de
datos: Scopus, Science Direct, EBSCOhost, Pubmed,
JSTOR, Red de Revistas Científicas de América Latina y
el Caribe, España y Portugal “Redalyc” y SpringerLink.
Se obtuvieron 5014 estudios primarios que mencionaran
la SG en vegetales. A través del gestor bibliográfico
Zotero se realizó la unificación de los estudios primarios
obtenidos en las siete bases de datos electrónicas,
así como la eliminación de estudios primarios
duplicados, excluyendo 779 estudios primarios.
Luego, se seleccionaron aquellos estudios que contenían
las palabras de búsqueda en el título (947 estudios
primarios). Seguidamente, se leyeron los resúmenes
de los trabajos que pasaron el filtrado del título y se
eligieron aquellos que tratasen la eficiencia de la SG
(se desecharon otros 358 estudios primarios). Los
estudios que pasaron la anterior etapa (589 trabajos),
fueron leídos de manera completa para seleccionar
aquellos estudios donde estaba publicada la información
necesaria para la construcción de la base de datos
(especies, carácter fenotípico, cantidad de marcadores,
métodos de estimación y la precisión de la predicción
(prediction accuracy), medida a través del coeficiente
de correlación “r” entre el fenotipo observado y los
valores de mejora predichos. Finalmente, la cantidad de
estudios primarios seleccionados para la conformación
de la base de datos fue de 68. En la Figura 1 se presenta
un diagrama de flujo de los pasos mencionados.
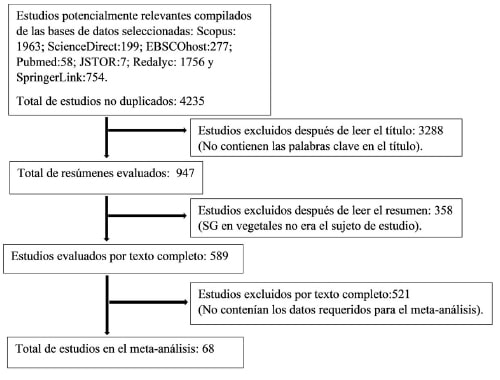
Figura 1. Diagrama de flujo de los criterios de exclusión para el meta-análisis.
Conformación de la base de datos para realizar el metaanálisis
La base de datos quedó conformada por 68 estudios
primarios, aunque en algunos de ellos se evaluaron
varios casos, i.e., más de un proceso de SG y por tanto
se pudieron obtener varios coeficientes de correlación,
totalizando 232 casos de SG. La base de datos conformada
a partir de los estudios primarios seleccionados contiene
la variable “Estudios” para identificar los estudios
primariosa través de sus autores y el año de la publicación.
Una columna denominada “Subgrupo_Especies” con
dos categorías: “cereales” (trigo, maíz, cebada, arroz
y centeno) y “otras especies” (césped, soja, colza,
remolacha, alfalfa, eucalipto, pera, raigrás, mandioca,
caña de azúcar y vid). Para este trabajo se consideraron
los datos provenientes de SG en cereales. La columna
“carácter” recopila la variable fenotípica analizada en
la población de entrenamiento (rendimiento de grano,
tiempo de floración, altura de planta, contenido de
proteína). La columna “Total_Marcadores” contiene
la cantidad de marcadores moleculares usados en
cada estudio primario y categorizada en terciles de la
distribución de cantidad de marcadores: baja≤1.700,
media=(1.700; 17.000] y alta>17.000. La columna
“Método_Estimación”, reporta los métodos de
estimación usados en la SG que luego son reagrupados
en dos categorías: “basados en BLUP” (BLUP, G-BLUP y
RR-BLUP) y “otros”, que incluyen “métodos bayesianos
o de aprendizaje automático” (M-BL, SVR, RKHS, Bayes
A y Bayes B). La última columna contiene el coeficiente
de correlación “r” entre el fenotipo observado y el
mérito genético predicho por SG reportado en cada
estudio primario. Un valor de correlación cercano a uno
indica alta eficiencia de la SG.
Los métodos basados en modelos mixtos y
consecuentemente en el mejor predictor lineal insesgado
(BLUP) han sido los más usados en SG.
El método
G-BLUP se caracteriza por asignar la misma varianza
a todos los loci, es decir, otorga la misma importancia
a cada alelo del marcador para obtener el predictor del
mérito genético, i.e., como suma los efectos alélicos
individuales; algunos marcadores pueden asociarse
a efectos nulos (Clark y van derWerf, 2013). Por otra
parte, el método RR-BLUP asume que todos los loci
tienen el mismo efecto, distinto de cero con las mismas
varianzas, pero esto no implica que todos los MM
tengan el mismo efecto. RR-BLUP estima la matriz de
relaciones entre los genotipos a partir de la información
provista por los MM, por lo tanto, algunos loci pueden
aportar al predictor y otros no. Los métodos bayesianos
ponderan el efecto de cada locus con distinta varianza a
diferencia de RR-BLUP. Entre los métodos basados en
aprendizaje automáticos, SVM ha sido uno de los más
usados en la SG, dado que al usar una función kernel
para los cálculos como el producto interno, resuelve el
problema de estimación con alta dimensionalidad. Por
ello, en estos tipos de modelos, la selección de la función
kernel se vuelve un factor clave, dado que la misma debe
reflejar la distribución característica de la muestra de
entrenamiento (Wang et al., 2018).
Meta-análisis
Se estimó la correlación (promedio ponderado de los
estudios) entre los valores observados y predichos por
el modelo en los estudios de SG realizados en maíz y
trigo; especies que representaron más del 50% del total
de casos detectados en la revisión sistemática. Se usó
un modelo de efectos aleatorios para el MA, ya que se
observó alta heterogeneidad entre estudios respecto a
los valores de r y la precisión reportada:
![]()
donde ri es la correlación observada, μ es la correlación
esperada entre el fenotipo observado y el mérito genético
predicho, τi es un efecto aleatorio asociado a cada estudio
primario que se supone con distribución N(0,τ) y i es un
término de error aleatorio con distribución N(0,σ2) que
mide la precisión dentro de cada caso de SG.
La heterogeneidad entre estudios se evaluó con el
estadístico I2, que permite cuantificar cuánto de la
variabilidad total en el estadístico de interés debe ser
atribuida a la variación entre estudios (Higgins et al.,
2003). Es una medida independiente del número de
estudios incluidos en el meta-análisis y de la unidad de
medida utilizada para cuantificar el efecto estudiado.
El estadístico I2, se expresa como una proporción, un
valor cercano a cero indica que la varianza observada
es espuria y por lo tanto, los estudios primarios
pueden considerarse homogéneos. Higgins et al. (2003)
sugirieron que valores de I2 hasta 25% podrían ser
indicadores de baja heterogeneidad, entre 25 y 50%
de mediana heterogeneidad y más de 75% de alta
heterogeneidad. Dado los altos valores encontrados para
I2, se llevaron a cabo análisis por subgrupos considerando
en cada análisis diferentes variables de clasificación,
como la cantidad de marcadores moleculares utilizados
(“Subgrupo_MM”) y el tipo de método de estimación
utilizado para la SG (variable “Método_Estimación”).
La estrategia de realizar análisis por subgrupo, además
de controlar la heterogeneidad entre estudios, permitió
detectar cómo estás variables contribuyen en la
estimación global de la eficiencia de la SG. Los metaanálisis
se realizaron con los datos transformados
a través del z de Fisher, pero los resultados de los
efectos globales fueron reportados en la métrica de
correlaciones. Los datos fueron analizados usando el
software R con el paquete meta (R Core Team 2020).
RESULTADOS
La SG en cereales ha convocado mayor atención
que en otras especies agrícolas por el acortamiento
aparejado en el ciclo de mejoramiento genético vegetal.
Probablemente, este hecho se asocie con la importancia
alimentaria de estas especies agrícolas que cuentan con
programas de mejoramiento genético vegetal en gran
parte del mundo.
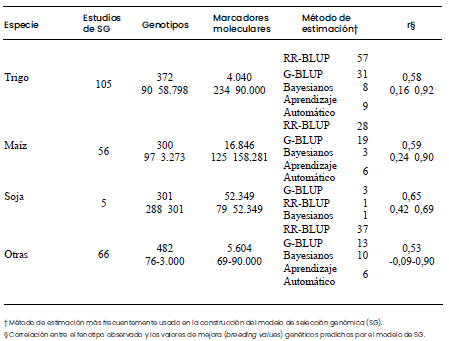
Los resultados reportados en la Tabla 1 indican
predominancia del uso de los métodos de estimación
RR-BLUP y G-BLUP en el ajuste del modelo estadístico
que permitirá obtener las predicciones para la SG. Es
importante destacar que se observó alta variabilidad
en los reportes de eficiencia de la SG; por ejemplo, en
trigo algunas publicaciones reportaban correlaciones
menores al 20% mientras que otras reportaban valores
de correlaciones en el valor fenotípico observado y el
valor genético predicho mayores al 80%. No obstante,
en la mayoría de los casos analizados la eficiencia de
la SG fue cercana al 60%, que en términos estadísticos
es mediana, pero en términos prácticos puede ser
suficiente.
Tabla 1. Estudios de selección genómica (SG) en cultivos (n=232) según especie, cantidad de genotipos y de
marcadores moleculares en la población de entrenamiento, método de estimación y eficiencia promedio.
La representación gráfica de los resultados se realiza con un Forest Plot (Figura 2), cuyas filas representan cada uno de los estudios primarios y la eficiencia de la SG en las especies trigo y maíz agrupadas según el método de estimación del modelo de SG (G-BLUP y RR-BLUP). El tamaño del efecto es la magnitud de la asociación entre el valor fenotípico observado y el valor genético predicho. Así, el gráfico permite visualizar la correlación de interés promedio (cuadrado) de cada estudio primario y su intervalo de confianza (IC) con nivel de confianza del 95%. Mientras menor es la amplitud del intervalo de confianza, mayor es la precisión en la estimación del coeficiente de correlación entre el valor fenotípico observado y el valor genético predicho. El cuadrado que representa el tamaño del efecto de cada estudio primario varía entre estudios para reflejar el peso de cada uno en la estimación del efecto global (correlación promedio ponderada). Un estudio con precisión relativamente buena, tendrá asignada mayor ponderación o peso para generar la estimación global. La precisión está gobernada por el tamaño de la muestra y por la varianza residual del estudio. Al final de la lista de estudios se visualiza el efecto global (rombo). Si la correlación global es estadísticamente distinta de cero, el valor de cría predicho por el modelo se correlaciona con el valor observado y la SG es eficiente. El efecto global de la correlación entre los valores observados y los valores predichos fue de 0,61, con un intervalo de confianza (IC) de [0,59-0,64] que confirma la eficiencia de la SG. La heterogeneidad entre estudios fue alta I2=99% y estadísticamente significativa p<0,001; como estrategia analítica para controlar parte de la heterogeneidad, se identificaron subgrupos relacionados al método de estimación (Figura 2) y a la cantidad de marcadores moleculares involucrados en la construcción del modelo de SG (Figura 3).
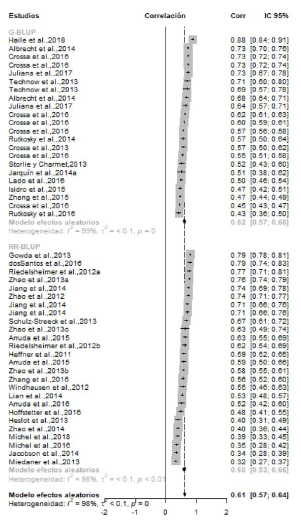
Figura 2. Forest Plot de la eficiencia de SG para los métodos de
estimación G-BLUP y RR-BLUP en trigo y maíz. El modelo de metaanálisis
ajustado fue un modelo de efectos aleatorios por subgrupos
(G-BLUP y RR-BLUP), contemplando de esta forma la heterogeneidad
entre estudios primarios y entre grupos. Las correlaciones se presentan
ordenadas de mayor a menor dentro de cada método de estimación.
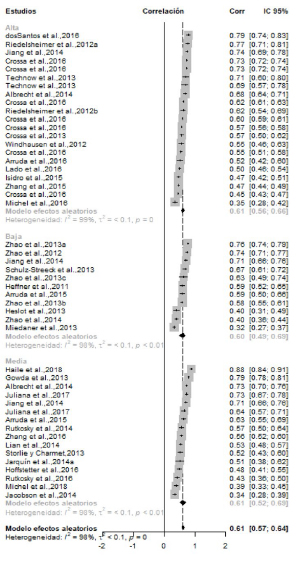
Figura 3. Forest Plot de la eficiencia de SG para distintas densidades
de marcadores moleculares categorizadas en: baja (menos de 1.700),
media (entre 1.700 y 17.000) y alta densidad de marcadores moleculares,
mayor a 17.000 para estudios primarios de trigo y maíz. El modelo
de meta-análisis ajustado fue un modelo de efectos aleatorios por
subgrupos de densidad de marcadores moleculares (Alta, Baja y
Media), contemplando de esta forma la heterogeneidad entre estudios
primarios y entre grupos. Las correlaciones se presentan ordenadas de
mayor a menor dentro de cada categoría de densidad de marcadores
moleculares.
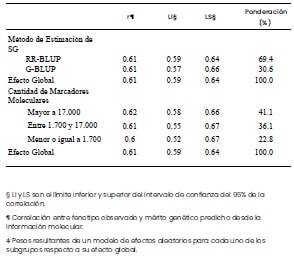
El intervalo de confianza (IC) para G-BLUP fue [0,57- 0,66] y para RR-BLUP [0,59-0,64]; en ambos casos no contienen al cero e indican que la eficiencia de la SG es similar entre ellos y que no estuvo condicionada por la selección de uno u otros métodos de estimación basados en BLUP. La superposición de los IC indica que no existen diferencias estadísticamente significativas entre ambos métodos de construcción del modelo para predecir mérito genético desde la información genómica. Los resultados del meta-análisis para trigo y maíz, realizado por subgrupos definidos por la densidad de marcadores moleculares (alta con más de 17.000 MM, media de 1.700 hasta 17.000 MM y baja con 1.700 o menos MM), mostraron similitud de la eficiencia alcanzada con distinta cantidad de marcadores moleculares (cerca del 60%) (Figura 3). Las ponderaciones o pesos reportados para las categorías alta y media fueron 41,1% y 36,1% respectivamente, mientras que las ponderaciones de la categoría de baja densidad de marcadores moleculares fue de 22,8%. Esto evidenció que la mayor contribución al efecto global de correlación entre los valores observados y predichos, se obtuvieron con densidades de marcadores moleculares medias y altas (Tabla 2).
Tabla 2. Estimación de la correlación entre fenotipo observado y mérito
genético predicho desde la información molecular de los métodos
de estimación más frecuentes de selección genómica y densidad de
marcadores moleculares en maíz y trigo (n=122).
DISCUSIÓN
La SG se comenzó a aplicar para la predicción genética
en animales (Meuwissen et al., 2001), no obstante,
fue adaptada rápidamente en los programas de
mejoramiento genético vegetal (Thavamanikumar et al.,
2015). Se presenta como una herramienta que permite
predecir de manera efectiva, los méritos genéticos de
los individuos a partir de información genómica, sin
necesidad de observar el fenotipo; así la selección se
puede realizar de manera más rápida.
Diferentes modelos estadísticos son usados para
asociar la variabilidad genómica a un carácter fenotípico
de interés y poder luego predecir el mérito genético de
un nuevo individuo. Sin embargo, han manifestado la
complejidad a la hora de comparar los resultados de
diferentes estudios, además consideran que no siempre
se ajusta el modelo más eficiente para un estudio en
particular (Wang et al., 2015). Los resultados del metaanálisis
conducido en este trabajo, sugieren que no existe
una diferencia significativa entre el ajuste del modelo
por G-BLUP o RR-BLUP, dos de los métodos más usados
para la construcción del modelo de SG.
Dado que los valores de correlación dependen
fuertemente de las varianzas de las variables que se
correlacionan, y que éstas pueden ser distintas bajo
distintos escenarios, el meta-análisis se realizó previa
transformación (z de Fisher) de las correlaciones
reportadas. La transformación del coeficiente de
correlación en la escala z de Fisher permite realizar el
meta-análisis en una escala común y luego transformar
el tamaño del efecto y los intervalos de confianza
logrados a las escalas originales (Borenstein et al.,
2009). Algunos autores sugieren que la transformación
z de Fisher podría producir un sesgo hacia la derecha
(Silver y Dunlap, 1987; Strube, 1988; Field, 2001; 2005).
Otros autores sugieren que la transformación logra
que el error estándar y, por lo tanto los intervalos de
confianza, dependan únicamente del tamaño muestral
y no del tamaño de la correlación observada, que puede
verse afectada por el error de muestreo (Silver y Dunlap,
1987; Field, 2005). Strube (1988) señaló que a medida
que aumenta el número de estudios primarios, el sesgo
debido a la transformacion es despreciable. En nuestro
trabajo el número de correlaciones derivadas de los
estudios primarios fue alto (n=232 cuando se trabajó con
todos los casos y n=48 cuando se analizaron las medias de
correlaciones por publicación). Mas allá de la existencia
de un posible sesgo, las correlaciones promedio, aunque
significativas, no fueron altas (aprox. r=0,6) sugiriendo
que existe espacio para mejorar la capacidad predictiva
de los modelos usados en SG en trigo y maíz.
Podría ser importante considerar efectos de interacción
entre los efectos de marcadores en los modelos lineales
usados para SG con el fin de incrementar la eficiencia de
la SG. Conceptualmente estos modelos son entendibles,
pero son computacionalmente difíciles de estimar
en la actualidad. Los métodos de base computacional
como algunos modelos de regresión de aprendizaje
automático podrían ofrecer una herramienta para el
tratamiento de interacciones de orden múltiple. Varios
métodos de ajustes de modelos de SG abordan los
problemas de la alta dimensionalidad y la complejidad
computacional capturando diferentes aspectos de la
asociación entre el genotipo y el fenotipo. Sin embargo,
el desempeño de diferentes métodos depende de la
arquitectura genética subyacente del carácter de interés
(Resende et al., 2012; Wang et al., 2018). Se ha discutido
en la literatura que la precisión de la SG depende
de la heredabilidad y de la distribución de los genes
causales (Desta y Ortiz, 2014) y consecuentemente
la heredabilidad del carácter en estudio se relaciona
positivamente a la precisión de la predicción. Otro factor
importante que debe ser considerado es la interacción
genotipo-ambiente (G×E); ya que predicciones sobre el
mismo carácter, pero evaluados en varios ambientes,
difieren considerablemente debido al efecto que causa
esta interacción (Wang et al., 2018). Su et al. (2012)
usaron predicción genómica con densidades extremas
de SNPs y observaron que cuando aumentaron la
cantidad de información genómica en un 1438%, la
precisión de la predicción aumentó solo en 0,5-1,0%.
Aun cuando se conoce que cuanto más marcadores
haya mejor será la predicción, la precisión es difícil de
mejorar significativamente cuando la densidad inicial de
marcadores ya es alta. Los resultados del meta-análisis
son consistentes con estas publicaciones. Nakaya e
Isobe (2012) realizaron otra revisión de estudios donde
se estimaba el mérito genético sobre bases de datos
simulados y bases de datos reales, y reportaron que la
predicción del valor de cría fue siempre mejor cuando
en los modelos se incorporó información provista por
marcadores moleculares distribuidos densamente en
el genoma respecto a los modelos tradicionales de la
construcción del BLUP solo con datos fenotípicos. En
otros estudios para caracteres relacionados a calidad de
grano en poblaciones biparentales de trigo, se observó
que la precisión de los modelos de SG alcanzó una
meseta con una densidad de marcadores moleculares
que se acercaba a los 256 (Heffner et al., 2011). Para
otras especies, como pinos, se observó que la máxima
capacidad predictiva se alcanzó en subconjuntos de 564
marcadores moleculares y luego disminuyó (Resende et
al., 2012). El mismo fenómeno se observó en otro estudio
pero con una densidad de marcadores mucho mayor,
ya que se usaron 3490 marcadores moleculares para
calibrar los modelos de SG (Oakey et al., 2016). Yang et
al. (2010) y Wang et al. (2017) concluyeron que cuando
la densidad de marcadores alcanza un cierto nivel, la
predicción genómica no se beneficia.
Entre los métodos estadísticos para ajustar el
modelo de SG, el más recomendado en la práctica
de mejoramiento, debido a su robustez y eficiencia
computacional es el G-BLUP (Wang et al., 2018).
No obstante, algunos algoritmos de aprendizaje de
máquinas, como RKHS (Reproducing Kernel Hilbert Space)
han tomado un papel muy importante en SG (Cuevas
et al., 2016). El método G-BLUP asume que todos los
efectos de marcador se distribuyen normalmente con
igual varianza (Meuwissen et al., 2001) y puede producir
resultados similares al método Bayes C (Ferrão et al.,
2017). En el trabajo de Gianola (2013) se aprecia que el
método de G-BLUP es similar al RR-BLUP, como también
se observó en el meta-análisis de este trabajo. Daetwyler et al. (2014) calibraron modelos de SG basados en G-BLUP
y en regresión bayesiana para predecir la resistencia a
la roya en trigo, y concluyeron que el modelo G-BLUP
presentó mejor desempeño que la regresión bayesiana.
En trigo, los modelos de SG bayesianos han mostrado
resultados prometedores en el contexto del análisis
de QTL (quantitative trait loci), siendo más sensibles al
aumento del número de QTL sin disminuir la precisión
en contextos de múltiples QTL (Wang et al., 2015).
La precisión que aportan los métodos G-BLUP
y RR-BLUP se puede mantener casi constante,
independientemente de la cantidad de QTL (Wang et
al., 2018). Si bien los modelos Bayes A y Bayes B han
mostrado buen desempeño (De los Campos et al., 2009) la
estimación de los modelos bayesianos puede llevar mucho
tiempo restringiendo su aplicación. Friedman et al. (2010)
mostraron una mejor alternativa con el algoritmo LASSO,
que logra un equilibrio entre la contracción selectiva de los
efectos alélicos y la eficiencia computacional. A pesar del
buen desempeño de los modelos bayesianos en SG animal
(Neves et al., 2014), Xu et al. (2017) señalaron que el modelo
G-BLUP tiene mejor desempeño que los bayesianos en la
predicción de caracteres relacionados con el rendimiento
en maíz y también, en la predicción del rendimiento en
grano de trigo. En el presente trabajo se observó que la
eficiencia de la SG no difiere significativamente entre
el uso de G-BLUP o RR-BLUP para el ajuste del modelo,
resultado consistente con lo publicado (Dong et al., 2016;
Ferrão et al., 2017).
En conclusión, aún cuando los modelos estadísticos
usados para SG en cultivos de importancia agrícola han
contribuido en la predicción genómica, permitiendo
seleccionar genotipos promisorios en etapas tempranas
de los programas de mejoramiento genético vegetal, aún
es necesario incrementar la capacidad predictiva. Los
modelos más usados en maíz y trigo para predecir mérito
genético han sido G-BLUP y RR-BLUP, sin diferencias
estadísticas en su performance. Si bien la capacidad
predictiva aumenta con el número de marcadores usados,
la respuesta no es lineal y en algunas situaciones el
incremento de estos podría no ser redituable en términos
del aumento producido sobre la eficiencia de la SG.
BIBLIOGRAFÍA
1. Akobeng A.K. (2005) Understanding systematic reviews and metaanalysis. Archives of Disease in Childhood 90 (8): 845-848.
2. Bhat J.A., Ali S., Salgotra R.K., Mir Z.A., Dutta S., Jadon V., Tyagi A., Mushtaq M., Jain N., Singh P.K., Singh G.P., Prabhu K.V. (2016) Genomic selection in the era of next generation sequencing for complex traits in plant breeding. Frontiers in Genetics 7: 221.
3. Borenstein M., Hedges L.V., Higgins J.P.T., Rothstein H.R. (2009) Introduction to meta-analysis. John Wiley & Sons. Ltd., Chichester, UK.
4. Borenstein M., Hedges L.V., Higgins J.P.T., Rothstein H.R. (2010) A basic introduction to fixed-effect and random-effects models for metaanalysis. Research Synthesis Methods 1 (2): 97-111.
5. Clark S.A., van der Werf J. (2013) Genomic best linear unbiased prediction (gBLUP) for the estimation of genomic breeding values. In: Gondro C., van der Werf J., Hayes B. (Eds.) Genome-Wide Association Studies and Genomic Prediction. Springer Protocols, pp. 321-330.
6. Cuevas J., Crossa J., Soberanis V., Pérez Elizalde S., Pérez Rodríguez P., de los Campos G., Montesinos López O.A., Burgueño J. (2016) Genomic prediction of genotype × environment interaction kernel regression models. The Plant Genome 9 (3).
7. Daetwyler H.D., Bansal U.K., Bariana H.S., Hayden M.J., Hayes B.J. (2014) Genomic prediction for rust resistance in diverse wheat landraces. Theoretical and Applied Genetics 127 (8): 1795-1803.
8. de los Campos G., Naya H., Gianola D., Crossa J., Legarra A., Manfredi E., Weigel K., Cotes J.M. (2009) Predicting quantitative traits with regression models for dense molecular markers and pedigree. Genetics 182 (1): 375-85.
9. Desta Z.A., Ortiz R. (2014) Genomic selection: genome-wide prediction in plant improvement. Trends in Plant Science 19 (9): 592-601.
10. Dong L., Xiao S., Wang Q., Wang Z. (2016) Comparative analysis of the GBLUP, emBayesB, and GWAS algorithms to predict genetic values in large yellow croaker (Larimichthys crocea). BMC Genomics 17 (1): 460.
11. Ferrão L.F.V., Ortiz R., Garcia A.A.F. (2017) Genomic selection: state of the art. In: Campos H., Caligari P. (Eds.) Genetic Improvement of Tropical Crops. Springer, pp. 19-54.
12. Ferreira González I., Urrútia G., Alonso Coello P. (2011) Revisiones sistemáticas y metaanálisis: bases conceptuales e interpretación. Revista Española de Cardiología 64 (8): 688-696.
13. Field A.P. (2001) Meta-analysis of correlation coefficients: a Monte Carlo comparison of fixed- and random-effects methods. Psychological Methods 6 (2): 161-180.
14. Field A.P. (2005) Is the meta-analysis of correlation coefficients accurate when population correlations vary? Psychological Methods 10 (4): 444-467.
15. Friedman J., Hastie T., Tibshirani R. (2010) Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of Statistical Software 33 (1): 1-22.
16. Gianola D. (2013) Priors in whole-genome regression: the Bayesian alphabet returns. Genetics 194 (3): 573-596.
17. Glass G.V. (1976) Primary, secondary, and meta-analysis of research. Educational Researcher 5 (10): 3-8.
18. Hawkins C., Yu L.X. (2018) Recent progress in alfalfa (Medicago sativa L.) genomics and genomic selection. The Crop Journal 6 (6): 565-575.
19. Heffner E.L., Jannink J.L., Iwata H., Souza E., Sorrells M.E. (2011) Genomic selection accuracy for grain quality traits in biparental wheat populations. Crop Science 51: 2597- 2606.
20. Heffner E.L., Sorrells M.E., Jannink J.L. (2009) Genomic selection for crop improvement. Crop Science 49 (1): 1-12.
21. Higgins J.P.T., Thompson S.G, Deeks J.J., Altman D.G. (2003) Measuring inconsistency in meta-analyses. BMJ 327 (7414): 557-560.
22. Meuwissen T.H., Hayes B.J., Goddard M.E. (2001) Prediction of Total Genetic Value Using Genome-Wide Dense Marker Maps. Genetics 157 (4): 1819-1829.
23. Nakaya A., Isobe S.N. (2012) Will genomic selection be a practical method for plant breeding? Annals of Botany 110 (6): 1303- 1316.
24. Neves H.H.R., Carvalheiro R., O’Brien A.M.P., Utsunomiya Y.T, do Carmo A.S, Schenkel F.S, Sölkner J., McEwan J.C., Van Tassell C.P., Cole J.B., da Silva M.V.G.B., Queiroz S.A., Sonstegard T.S., Garcia J.F. (2014) Accuracy of genomic predictions in Bos indicus (Nellore) cattle. Genetics Selection Evolution 46 (1): 17.
25. Oakey H., Cullis B., Thompson R., Comadran J., Halpin C., Waugh R. (2016) Genomic selection in multi-environment crop trials. G3: Genes, Genomes, Genetics 6 (5): 1313- 1326.
26. Pai M., McCulloch M., Gorman J.D., Pai N., Enanoria W., Kennedy G., Tharyan P., Colford J.M. (2004) Systematic reviews and meta-analyses: an illustrated, step-by-step guide. The National Medical Journal of India 17 (2): 86-95.
27. R Core Team (2020) R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna.
28. Resende M.F.R., Muñoz P., Resende M.D.V., Garrick D.J., Fernando R.L., Davis J.M., Jokela E.J., Martin T.A., Peter G.F., Kirst M. (2012) Accuracy of genomic selection methods in a standard data set of Loblolly Pine (Pinus taeda L.). Genetics 190 (4): 1503-1510.
29. Sánchez Meca J. (2010) Cómo realizar una revisión sistemática y un meta-análisis. Aula Abierta 38 (2): 53-64.
30. Silver N.C., Dunlap W.P. (1987) Averaging correlation coefficients: should Fisher’s z transformation be used? Journal of Applied Psychology 72 (1): 146-148.
31. Strube M.J. (1988) Averaging correlation coefficients: influence of heterogeneity and set size. Journal of Applied Psychology 73 (3): 559-568.
32. Su G., Brøndum R.F., Ma P., Guldbrandtsen B., Aamand G.P., Lund M.S. (2012) Comparison of genomic predictions using mediumdensity (~54,000) and high-density (~777,000) single nucleotide polymorphism marker panels in Nordic Holstein and Red Dairy Cattle populations. Journal of Dairy Science 95 (8): 4657-4665.
33. Thavamanikumar S., Dolferus R., Thumma B.R. (2015) Comparison of genomic selection models to predict flowering time and spike grain number in two hexaploid wheat doubled haploid populations. G3: Genes, Genomes, Genetics 5 (10): 1991-1998.
34. Voss Fels K.P., Cooper M., Hayes B.J. (2019) Accelerating crop genetic gains with genomic selection. Theoretical and Applied Genetics 132 (3): 669-686.
35. Wang X., Li L., Yang Z., Zheng X., Yu S., Xu C., Hu Z. (2017) Predicting rice hybrid performance using univariate and multivariate GBLUP models based on North Carolina mating design II. Heredity 118: 302-310.
36. Wang X., Xu Y., Hu Z., Xu C. (2018) Genomic selection methods for crop improvement: Current status and prospects. The Crop Journal 6 (4): 330-340.
37. Wang X., Yang Z., Xu C. (2015) A comparison of genomic selection methods for breeding value prediction. Science Bulletin 60 (10): 925-935.
38. Wu X.L., Hu Z.L. (2012) Meta-analysis of QTL mapping experiments. In: Rifkin S.A. (Ed.) Quantitative Trait Loci (QTL): Methods and Protocols. Totowa, NJ, Humana Press, pp. 145-171.
39. Xu Y., Xu C., Xu S. (2017) Prediction and association mapping of agronomic traits in maize using multiple omic data. Heredity 119 (3): 174-184.
40. Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W., Goddard M.E., Visscher P.M. (2010) Common SNPs explain a large proportion of the heritability for human height. Nature Genetics 42: 565-569.