


Vol. XXXI Issue 1
Article 4
ARTÍCULOS ORIGINALES
Identificación de alteraciones moleculares en pacientes Venezolanos con diagnóstico de leucemia linfoblástica aguda
Identification of molecular alterations in Venezuelan patients with acute lymphoblastic leukemia diagnosis
Castro Y.C.1, Utrera R.1
1 Laboratorio Genética Molecular
Humana, Universidad Simón Bolívar,
Venezuela.
Corresponding author:
Yarlenis Castro
liceyarlenis@gmail.com
DOI: 10.35407/bag.2020.31.01.04
Received: 08/07/2019
Revised version received: 04/23/2020
Accepted: 04/28/2020
RESUMEN
La Leucemia Linfoblástica Aguda (LLA) es la neoplasia más frecuente en edad pediátrica. En los últimos años, entre el 15 y 20% de los pacientes fracasan en el tratamiento. Conocimientos en citogenética y biología molecular repercuten de manera importante en la determinación del pronóstico y del esquema de tratamiento adecuado. En Venezuela existe un conocimiento limitado en cuanto a la genética molecular de esta alteración onco-hematológica. El objetivo del trabajo fue evaluar las alteraciones genéticas más frecuentes en pacientes venezolanos con diagnóstico clínico de leucemia linfoblástica aguda. Se realizó un estudio transversal, descriptivo y prospectivo de 2006 a 2014, en el que se evaluaron las translocaciones ETV6/RUNX1, MLL/AF4, TCF3/PBX1, BCR/ABL1, así como las mutaciones en los genes PAX5 y FLT3 mediante el uso de diferentes tipos de PCR. Ciento treinta pacientes con diagnóstico clínico de leucemia linfocítica aguda fueron incluidos en el estudio. Se identificaron alteraciones moleculares en 56 pacientes (43,1%), en los que observamos la presencia de una o varias alteraciones en conjunción en un mismo paciente. Las alteraciones identificadas fueron t(12;21) (11,5%), t(4;11) (8,5%), t(1;19) (10%), t(9;22) (20,8%), ITD-FLT3 (14,8%), mutación P80S (4,2%) y S77del (4,2%) en el gen PAX5. La prevalencia de BCR/ ABL, es una de las más altas que ha sido descrita hasta ahora en casos de LLA donde la mayor parte de la población está conformada por pacientes pediátricos. Estos resultados representan el primer estudio molecular de la LLA en Venezuela, sentando las bases para el diagnóstico y seguimiento de la enfermedad en su población.
Palabras clave: Leucemia Linfoblástica Aguda; Translocaciones; ETV6/RUNX1; MLL/AF4; TCF3/PBX1; BCR/ABL1; PAX5; FLT3.
ABSTRACT
Acute Lymphoblastic Leukemia (ALL) is the most common neoplasm in pediatric age. In recent years, between 15 and 20% of patients failed in their treatments. Knowledge on cytogenetics and molecular biology has an important impact on the determination of the prognosis and the appropriate treatment scheme. In Venezuela there is limited knowledge regarding the molecular genetics of this onco-hematological alteration. The aim of this work was to evaluate the most frequent genetic alterations in Venezuelan patients with a clinical diagnosis of acute lymphoblastic leukemia. A cross-sectional, descriptive and prospective study was carried out from 2006 to 2014, in which the translocations ETV6/RUNX1, MLL/AF4, TCF3/PBX1, BCR/ABL1, as well as mutations in the PAX5 and FLT3 genes were evaluated through the use of different types of PCR. One hundred and thirty patients with a clinical diagnosis of acute lymphocytic leukemia were included in the study. Molecular alterations were identified in 56 patients (43.1%), in which we observed the presence of one or several alterations in conjunction in the same patient. The alterations identified were t(12; 21) (11.5%), t(4; 11) (8.5%), t(1; 19) (10%), t(9; 22) (20.8%), ITD-FLT3 (14.8%), P80S mutation (4.2%) and S77del (4.2%) in the PAX5 gene. The prevalence of BCR/ABL is one of the highest described so far in cases of ALL where most of the population is made up of pediatric patients. These results represent the first molecular study of ALL in Venezuela, laying the foundations for the diagnosis and monitoring of the disease in its population.
Key words: Acute Lymphoblastic Leukemia; Translocations; ETV6/RUNX1; MLL/AF4; TCF3/PBX1; BCR/ABL1; PAX5; FLT3.
INTRODUCCIÓN
La Leucemia Linfoblástica Aguda (LLA), se define
clínicamente como la expansión clonal de células
progenitoras linfoides (linfoblastos), comprometidas a
un linaje, ya sea B o T, transformadas malignamente a
través de un bloqueo en un punto de su diferenciación,
dando lugar a células inmaduras e indiferenciadas.
Estas células inmaduras que se multiplican de forma
incontrolada, infiltran la médula ósea, producen un
grado variable de pancitopenia y pueden comprometer
diferentes órganos y/o sistemas y causar la muerte por
hemorragia y/o infección (Teitell y Pandolfi, 2009).
Se caracteriza por tener una distribución bimodal con
un primer pico en pacientes menores de 20 años (±60%)
y el segundo a partir de los 45 años de edad (20%) (Smith
et al., 1999). Representa el 75-80% de las leucemias
agudas en edad pediátrica con un pico de incidencia
entre los 2 a 5 años y un 20% en edad adulta (Smith et al.,
1999; Wiemels, 2012).
Venezuela es un país con una población aproximada
de 33 millones de habitantes, y el cáncer constituye la
segunda causa de muerte según el Anuario de Mortalidad
2014, publicado en el año 2015 por el Ministerio del
Poder Popular para la Salud. En los registros oficiales,
que datan del año 2009, donde se engloban todos los
tipos de cáncer sin hacer diferenciación entre ellos, se
indicó que el cáncer fue causante de 20.288 defunciones,
representando el 15,5% del total registrado. Los datos
para el año 2019, mostraron un incremento en la
cifra de fallecidos por cáncer del 16,6% con respecto
a lo establecido en las proyecciones oficiales. Esta
posición en la que se localiza el cáncer en Venezuela
se ha mantenido en los últimos 25 años, y sólo ha sido
superada por enfermedades del corazón. Aun cuando
en Venezuela no existen cifras concretas, las tasas de
morbi-mortalidad asociadas con la leucemia en el país
son cada vez mayores (Villalta et al., 2019; Capote, 2012).
Hasta hace aproximadamente 45 años la LLA era
una enfermedad incurable; pero desde la década
del 70 se abrieron nuevas perspectivas, motivado al
reconocimiento de una serie de factores que produjeron
cambios en el manejo de la enfermedad, como la
necesidad de dar profilaxis al sistema nervioso central
y el uso de protocolos con poliquimioterapia que, en
su conjunto, resultaron en una mejoría de las tasas de
curación en muchos países del mundo. Actualmente,
utilizando las mismas drogas de manera más apropiada,
países desarrollados reportan una tasa de curación entre
el 80 y el 85% y algunos centros internacionales de
tratamiento e investigación reportan hasta de un 90%
(Globocan, 2012; Tirado et al., 2007). En nuestro país la
situación se presenta de forma diferente. A pesar que la
respuesta inicial se obtiene en el 85% de los casos, la
tasa de recaídas medulares es alta (13-15%) (Landolfi
et al., 2013). Esto puede deberse a posibles fallas en el
tratamiento de quimioterapia de inducción o a factores
inherentes a alteraciones moleculares adquiridas luego
de la remisión inicial (Landolfi et al., 2013).
En los últimos años, gracias a la citogenética y a la
biología molecular, se han efectuado grandes avances
en el conocimiento de los mecanismos que determinan
la transformación maligna de las células precursoras de
la hematopoyesis. Estos conocimientos se han traducido
en un aumento de los métodos de diagnóstico, terapia
molecular dirigida, así como también, mejoras en el
seguimiento y pronóstico de la enfermedad (Pui et al.,
2008; 2011).
Se estima que en el 60% de las leucemias agudas existe
una alteración cromosómica como las aneuploidías,
rearreglos estructurales, translocaciones, inversiones,
deleciones, monosomías y trisomías (Pui et al., 2011).
La identificación de estas alteraciones moleculares
específicas es actualmente un elemento indispensable
para la estratificación de las leucemias en distintos grupos
de pronóstico para su tratamiento adecuado. Asimismo,
se sabe que las alteraciones genéticas de las neoplasias
son generalmente los agentes causales de la enfermedad,
y definen distintos comportamientos biológicos que
se traducen en diferentes comportamientos clínicos y
finalmente, en pronósticos muy variables (Abdullaev et
al., 2000).
El análisis detallado de los rearreglos citogenéticos
en las leucemias, por varios años ha aportado
información importante para aclarar las incidencias de
anormalidades individuales y su significado pronóstico
(Jiménez Morales, 2002).
A pesar de los grandes avances que han ido
apareciendo en la sociedad referidos a nuevos
conocimientos y tratamientos de las leucemias, todavía
fallece un porcentaje considerable de niños debido a esta
enfermedad o por los efectos secundarios de la terapia;
por lo tanto, la leucemia sigue siendo la principal causa
de muerte por padecimientos oncológicos en pediatría,
razón por la cual en la actualidad se están realizando
diversos estudios enfocados a determinar la frecuencia
y prevalencia de estas alteraciones en las diferentes
poblaciones, así como en supervisar y analizar las
diversas anormalidades genéticas presentes en las
células leucémicas (Pulte et al., 2009).
El conocimiento aún limitado en relación a la genética
molecular de la leucemia en pacientes pediátricos
venezolanos, pudiera ser la causa de que en algunos
niños sus tratamientos sean sobredimensionados, o por
el contrario, subestimados. En efecto, el uso de esquemas
de quimioterapia muy agresivos aumenta el riesgo de
desarrollar neoplasias secundarias, y con ello mayores
dificultades en estos pacientes (Díaz Beveridge y Urtasun
Aparicio, 2003). Por ello, se ha hecho necesario el
desarrollo y/o mejoramiento de técnicas de diagnóstico
más eficientes, como por ejemplo, la detección de
marcadores moleculares asociados a pronóstico.
La detección y conocimiento de los marcadores
moleculares frecuentes en la LLA como son ETV6/RUNX1,
BCR/ABL, TCF3/PBX1 y MLL/AF4 permitirá corroborar el
diagnóstico, establecer la gravedad de la enfermedad
y contribuir en el desarrollo de nuevos esquemas de
tratamiento; los cuales a su vez, permitirán establecer
terapias más adecuadas e individualizadas basadas
en las características genéticas propias de las células
malignas presentes. También permitirá monitorear los
procesos de remisión o agravamiento de la enfermedad,
para finalmente aumentar la sobrevida y calidad de vida
de los pacientes con dicha patología (Woo et al., 2014).
Un punto interesante en este tipo de estudio es
que los individuos responden de manera diferente
a las drogas, y que a veces los efectos pueden ser
impredecibles. Las diferencias en la secuencia del ADN
que altera la expresión o función de las proteínas a
las que van dirigidas estas drogas, puede contribuir
significativamente a la variación en la respuesta de
los individuos. Por ejemplo, mientras los niños con la
t(12;21) ETV6/RUNX1 responden bien a terapias basadas
en antimetabolitos, los que presentan la t(9;22) BCR/
ABL no lo hacen de igual manera y en su defecto deben
ser tratados con inhibidores de tirosin-kinasa (Pui et
al., 2011). Por tal motivo, este estudio molecular de las
distintas alteraciones que conducen a la transformación
maligna de las células linfoides preparará el camino
para el desarrollo de terapias individualizadas dirigidas
al defecto genético causante de la proliferación anómala
que presenten menores efectos secundarios, evitando
así tratamientos excesivos e ineficientes y optimizando
de esta manera el tratamiento.
El objetivo de este estudio fue investigar los eventos
moleculares más frecuentes, entre ellos, ETV6/RUNX1,
BCR/ABL, TCF3/PBX1 y MLL/AF4, y las alteraciones en
los genes PAX5 y FLT3, relacionados a la Leucemia
Linfocítica Aguda en pacientes venezolanos con edades
comprendidas entre 0 y 66 años (abarcando los grupos
etarios más afectados por esta alteración hematológica)
diagnosticados clínicamente con LLA.
MATERIALES Y MÉTODOS
Se realizó un estudio transversal, descriptivo y prospectivo, efectuado entre los años 2006 al 2014, con muestras tomadas bajo previo consentimiento informado, llevado a cabo en forma conjunta entre el Laboratorio de Genética Molecular Humana de la Universidad Simón Bolívar (USB), el Hospital “Miguel Pérez Carreño”, Hospital “J.M de los Ríos” y el Banco Municipal de Sangre de Caracas, Venezuela.
Pacientes
Se evaluaron 130 pacientes con diagnóstico clínico y
citomorfológico de LLA, con edades comprendidas entre
0 y 66 años, abarcando ambos géneros. La toma de
muestra de los pacientes se llevó a cabo en el servicio de
hematología de los centros de salud antes mencionados.
Las muestras procesadas fueron obtenidas a través de
aspirados de médula ósea (MO) y/o sangre periférica
(SP), colectadas en tubos con anticoagulante EDTA,
mantenidas a temperatura ambiente. Se lograron
recolectar datos de laboratorio en 89 pacientes;
asimismo, en sólo 38 órdenes de las 130 se denotó alguna
observación donde se indicó si el paciente se encontraba
para diagnóstico inicial o si estaba en recaída medular
luego de remisión o tratamiento.
Métodos de laboratorio
Extracción de ARNtotal (ARNt) y síntesis de
ADNcomplementario (ADNc) por transcripción reversa
Los leucocitos (glóbulos blancos) se obtuvieron
mediante el método de separación por gradiente de
Ficoll, siguiendo las instrucciones sugeridas por la casa
comercial “Amersham Biosciences”. El concentrado de
linfocitos se almacenó en Trizol®Reagent (Invitrogen) a
-80 °C hasta su procesamiento. La extracción de ARN se
realizó según el método de Chomczynski (Chomczynski et al., 1987).
La integridad del ARNt se evaluó tanto por
cuantificación espectofotométrica donde además se
determinó la pureza, estimando el índice de absorbancia
260:280 nm, así como a través de electroforesis en geles
de agarosa bajo condiciones desnaturalizantes (Agarosa
UltraPure TM RNAsafree, Invitrogen) al 1%, MOPS 10X
(MOPS 0,2 M, acetato de sodio 0,05M, EDTA 0,01M, pH
8,0 y formaldehído 37%), obteniendo de esta manera
una estimación visual aproximada de la concentración
del ARNt. Los geles seleccionados fueron fotografiados
y visualizados empleando el equipo y programa “Gel Doc
2000TM Gel Documentation System CCIR”, Quantity One 4.2.1, ambos de BIO RAD.
A partir del ARN mensajero se obtuvo un ADN
complementario mediante la reacción de transcriptasa
reversa (enzima M-MVL Invitrogen), el cual se usó
en cada uno de los casos para amplificar secuencias
específicas de las alteraciones moleculares evaluadas.
Reacción de PCR
El ADNc previamente sintetizado se utilizó como
molde para las siguientes reacciones de amplificación
utilizando en común componentes como: buffer de
enzima (Tris-HCL 10 mM pH83, KCL 50 mM) 1X, dNTP´s
0,2 mM, MgCl22 mM, TaqADN polimerasa 0,05 U/μL
y cebadores 0,75 μM. Todos estos ensayos de PCR se
llevaron a cabo en un termociclador programable Mod
TC-100TM (MJ Research Inc.).
a) Amplificación del gen GAPDH mediante PCR
convencional (PCR-I) para evaluar la integridad de
los ADNc sintetizados. La secuencia de los cebadores
utilizados GAPDHup y GAPDHdown se pueden observar
en la tabla 1; las condiciones de la pcr: 35 ciclos 95 °C- 4
min, 95 °C- 30 seg, 50 °C- 1 min, 72 °C- 1 min.
b) Amplificación por PCR anidada (PCR-I, PCR-II) para
identificar las quimeras ETV6/RUNX-1, MLL/AF4, E2A/
PBX-1 y amplificación por PCR convencional para la
identificación de la quimera BCR/ABL y sus variantes más
frecuentes en la LLA (p190 y p210). La secuencia de los
cebadores utilizados tanto en PCR-I como PCR-II, según
el orden en que se nombraron las quimeras evaluadas
se observan en la tabla 1. En cuanto a las condiciones,
particularmente para la detección de la quimera ETV6/
RUNX-1, en la PCR-I se llevó a cabo una PCR con
condiciones características de una PCR “Touch Down”,
la cual se llevó a cabo en dos etapas. En la primera etapa
se realizó un ciclo a 94 °C 3 min, 8 ciclos de hibridación
a 50 °C 45 seg donde por cada ciclo se disminuyó 1°
C la temperatura y una extensión a 72 °C 45 seg. En la
segunda etapa se realizaron 35 ciclos 94 °C 45 seg, 60
°C 45 seg, 72 °C 1 min y extensión final 72 °C 3 min. Para
el resto de las translocaciones, las condiciones solo
variaron en la temperatura de hibridación 60 °C (MLL/
AF4), 65 °C (TCF3/PBX1) y 55 °C (BCR/ABL1) y el tiempo
de extensión final para BCR/ABL que fue de 10 min, para
las otras quimeras fue de 4 min.
c) Amplificación por PCR convencional para la detección
de duplicaciones en tándem en los exones 14 y 15 del gen
FLT3 y PCR-RFLP para la detección de la mutación D835
en el exón 20 del mismo gen. Para la ejecución del último
ensayo mencionado, posterior a la amplificación por
PCR se llevó a cabo la digestión del producto amplificado
con la enzima EcoRV (Promega). Se amplificaron las
muestras de 54 del total de pacientes evaluados con
diagnóstico clínico y citomorfológico de LLA. Dentro de
estas 54 muestras escogidas, estuvieron todas aquellas
que resultaron ser positivas para la translocación MLL/
AF4, esto debido a que una de las características que
distingue a las LLA generadas por alteraciones del gen
MLL es la alteración del gen FLT3 (Armstrong et al.,
2002; 2003; Stam et al., 2005). El resto de los pacientes
se distribuyeron de la siguiente manera: pacientes
positivos para el resto de las translocaciones evaluadas
ETV6/RUNX-1, TCF3/PBX1, BCR/ABL-1 y pacientes cuya
muestra no amplificó para ninguna de las alteraciones
cromosómicas antes mencionadas. Para el ensayo de
detección de duplicaciones en tándem se utilizó una
concentración final de MgCl2de 1Mm y los productos
de PCR obtenidos fueron analizados mediante corridas
electroforéticas de geles de agarosa al 3%, para garantizar
la visualización de las bandas correspondientes a los
fragmentos presentes de las ITD. La secuencia de los
cebadores utilizados se presentan en la tabla 1, mientras
que las condiciones de la pcr fueron las mismas para las
dos amplificaciones, 35 ciclos 94 °C 4 min, 95 °C 30 seg,
51 °C 1 min, 72 °C 1 min y extensión final por 7 min.
d) AS-PCR para la detección de la mutación puntual P80R
en el gen PAX-5. Para este ensayo solo se analizaron
24 muestras de pacientes con diagnóstico clínico y
citomorfológico de LLA, debido a que al momento de
realizar el análisis de esta mutación, la cantidad disponible
de muchas de las muestras de pacientes evaluados era
insuficiente. Para ello, fueron seleccionados pacientes
positivos de cada una de las translocaciones evaluadas
previamente: ETV6/RUNX-1, MLL/AF4, TCF3/PBX-1, BCR/
ABL-1 y pacientes cuyas muestras no amplificaron para
ninguna de las alteraciones cromosómicas evaluadas. La
secuencia de los cebadores se observan en la tabla 1 y las
condiciones de pcr: 95 °C 4 min, 95 °C 1 min, 55 °C 1 min,
72 °C 1 min, 72 °C 7 min durante 35 ciclos.
Tabla 1. Secuencia de cebadores para amplificación por PCR

Los productos de PCR fueron visualizados en gel de agarosa al 1,5%, teñidos con bromuro de etidio. Todas las reacciones contaron con controles positivos y negativos. Asimismo, los productos de PCR fueron secuenciados utilizando el servicio comercial de Macrogen Inc. Korea y se corrió BLAST para la coincidencia nucleotídica (http://www.ncbi.nlm.gov/cgibin/BLAST/nph-blast). Para cambiar la secuencia a reverso complementario en los casos donde se tuvo que secuenciar con el Oligo antisentido se utilizó el software en línea http://www. bioinformatics.org/sms/rev_comp.html; para realizar alineamientos y traducción de secuencias se utilizó y http://bio.lundberg.gu.se/. Además se utilizaron los software SIFT (http://sift.jcvi.org/) y PROVEAN (http:// provean.jcvi.org/) para determinar si las mutaciones encontradas podrían ejercer un efecto negativo en la función de la proteína afectada, y predecir si la sustitución o deleción de un aminoácido tenía un impacto en la función biológica de la proteína.
RESULTADOS
Se evaluó la integridad, tanto de los ARNt extraídos como
del ADNc sintetizado de las 130 muestras de pacientes
con diagnóstico clínico y citomorfológico de leucemia
linfocítica aguda (LLA), para confirmar si cumplían
con la cantidad de ARN necesario para el proceso de
análisis de las alteraciones moleculares. Se encontró
que en algunos casos, la cantidad de ARNt obtenida fue
baja, sin embargo esto no afectó los posteriores análisis
realizados sobre las muestras. Aún en los casos de
muestras que presentaron una degradación parcial fue
posible realizar los ensayos posteriores por PCR, ya que
los fragmentos amplificados se encontraban delimitados
en un promedio de tamaño comprendido entre 200/800
pb. Por otra parte, en los casos que contenían abundante
cantidad de ARNt, debido a la cuantiosa cantidad de
células inmaduras, se procedió a la preparación de las
diluciones respectivas para así evitar que las reacciones
de amplificación posteriores se vieran inhibidas por
exceso de muestra.
De 130 pacientes, 86 eran varones. El grupo más
frecuente de edad al diagnóstico fue de 0 a 10 años, 89
pacientes (68,5%); seguido del grupo de 11 a 18 años,
31 pacientes (23.8%). El grupo de menor incidencia
lo ocuparon pacientes con edad mayor a 18 años, 10
pacientes (7.7%).
El promedio del total de los pacientes según
presentación del gen de fusión u otra alteración génica
se presenta en la Tabla 2.
Tabla 2. Características de los pacientes según presentación del gen de fusión u otra alteración génica
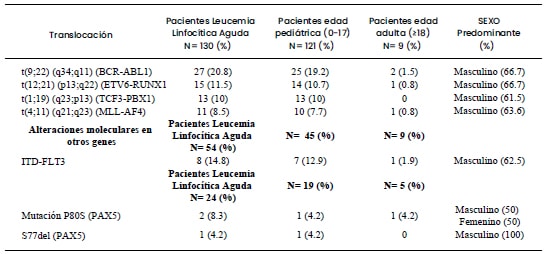
Alteraciones moleculares evaluadas
A) Translocación ETV6/RUNX1: Se detectó este transcrito
en 15/130 (11,5%) pacientes evaluados. En el subgrupo
correspondiente a los pacientes en edad pediátrica
comprendida entre 0 y 18 años, se encontró la
translocación ETV6/RUNX1 en 14/15 (93,3%) de ellos.
Mientras que en la población adulta se halló positividad
para esta translocación en 1/15 (6,7%), paciente
femenino de 66 años de edad. La edad promedio de los
pacientes pediátricos positivos para ETV6/RUNX1 (14) fue
de 8 años, de los cuales 10/14 (71,4%) fueron masculinos
y 4/14 (28,6%) fueron del género femenino.
Doce de las muestras evaluadas positivas para la
quimera ETV6/RUNX1, presentaron el mismo punto de
corte descrito y señalado en los estudios realizados en
otros países en relación a la translocación ETV6/RUNX1,
el cual incluyó la unión del extremo 3´ del exón 5 del
gen ETV6, con el extremo 5´ del exón 3 del gen RUNX1
(Paciente A-Figura 1). Contrariamente, 3 de las muestras
de los pacientes con LLA evaluados presentaron un
punto de corte distinto al clásicamente señalado en la
bibliografía. En este último caso, el punto de corte se
ubicó entre el exón 5 del gen ETV6 y el exón 4 del gen
RUNX1 (Paciente B- Figura 1). Este nuevo punto de corte
encontrado en estos pacientes venezolanos, no ha sido
aún reportado en las investigaciones de otros países.
B) Translocación MLL/AF4: Se detectó esta alteración en
11/130 (8,5%) del total de pacientes evaluados. La edad
promedio fue de 6,1 años, de los cuales 7/11 (63,64%)
fueron del sexo femenino y 4/11 (36,36%) del sexo
masculino. Del total de pacientes pediátricos que se
evaluaron en este estudio (120 - 92,3%), se encontró
la translocación MLL/AF4 en 10 (8,3%) de ellos, con
edades comprendidas entre 0 y 12 años. Mientras que
de la población adulta fueron evaluadas 10 muestras de
pacientes con LLA (7,7%), detectando positividad para
esta translocación en un paciente femenino (10%), de 19
años de edad.
En todas las secuencias obtenidas de las muestras
positivas para esta translocación se verificó la posición
del punto de corte, el cual estuvo ubicado entre el final
del exón 8 del gen MLL y el inicio del exón 5 del gen AF4, lo cual representa el punto de corte descrito en la
literatura para la translocación MLL/AF4. Al realizar el
análisis in silico, en lo que respecta a la traducción de
secuencia para cada una de las muestras analizadas, se
observó que se mantuvo el marco de lectura, y por ende,
se llevó a cabo la traducción de la proteína quimérica
MLL/AF4 en todos los casos detectados y evaluados.
C) Translocación BCR/ABL1: Este transcrito fue detectado
en 27/130 (20,8%) muestras de pacientes con diagnóstico
clínico de LLA, de las cuales 22 (16,9%) fueron positivas
solo para la variante p190 de la translocación BCR/ABL,
2 (1,5%) fueron positivas solo para la variante p210
de la translocación BCR/ABL, y 3 (2,3%) presentaron
ambas variantes (p190 y p210) simultáneamente. La
edad promedio de los pacientes con esta translocación,
independientemente de la variante presente fue de
10 años, donde el sexo masculino fue prevalente,
encontrándose 18/27 (66,7%) pacientes positivos del
sexo masculino; mientras que el sexo femenino por
su parte representó el 33,3% (9/27 pacientes) de las
muestras positivas encontradas para la translocación BCR/ABL. Haciendo énfasis en la edad y género de los
pacientes positivos encontrados para cada variante, se
obtuvo que para la variante p190 (e1a2) la edad promedio
fue de 10,4 años, encontrándose 17 pacientes de sexo
masculino (68%) y 8 pacientes de sexo femenino (32%).
Por su parte, de los 5 pacientes positivos para la variante
p210, la edad promedio fue de 15,8 años donde el sexo
masculino prevaleció representando el 80%, mientras
que el sexo femenino solo representó el 20% del total de
muestras positivas para esta variante.
En lo que respecta a las muestras positivas para la
variante p190, en todas las secuencias se observó que
la posición del punto de corte estuvo ubicado entre
el exón 1 del gen BCR y el exón 2 del gen ABL1, lo cual
representa el punto de corte descrito en la literatura para
la translocación BCR/ABL variante p190. Por su parte,
en la secuencia de todas las muestras positivas para la
variante p210, se obtuvo un punto de corte generado por
la unión del exón 13 del gen BCR con el exón 2 del gen ABL1 (variante b2a2).
D) Translocación TCF3/PBX1: Se detectó esta translocación
en 13/130 (10%) del total de pacientes analizados,
mostrando la mayoría de las muestras positivas una
banda con un tamaño aproximado de 289 pb (9/13),
mientras que 4/13 pacientes positivos mostraron una
banda de 200 pb aproximadamente. De las 13 muestras
de pacientes positivos, 8/13 (61,5%) fueron del sexo
masculino, mientras que 5/13 (38,5%) fueron del sexo
femenino. Todos los pacientes positivos encontrados en
este estudio para esta translocación pertenecen al grupo
pediátrico con edades comprendidas entre 3 y 13 años
con un promedio de edad de 6 años.
Post-secuenciación se observó que la porción de la
secuencia que fue reconocida correspondió al exón 3 del
gen PBX1. Destacando que el cebador con el que se envió a
secuenciar fue el correspondiente al gen TCF3 en segunda
ronda (TCF3.2), el cual se encuentra ubicado en el exón
14 del gen TCF3. Por lo tanto, se dedujo que la ausencia
de región perteneciente al gen TCF3 en la secuencia de
los pacientes positivos para la translocación TCF3/PBX1,
pudo deberse a que el cebador (TCF3.2) con el cual fueron
enviadas a secuenciar estas muestras, se encuentra
ubicado a solo 7 bases nucleotídicas del extremo 3´ del
exón 14. Por lo que, al encontrarse cercano al final del
exón había pocas probabilidades de detectar parte de
la secuencia del gen TCF3. De esta manera, la posición
del punto de corte para las muestras positivas para esta
alteración, estuvo ubicado entre el exón 14 del gen TCF3 y 7 el exón 3 del gen PBX1.
E) Alteraciones en el gen FLT3: Para el análisis de
detección de las duplicaciones internas en tándem
(ITD), se evaluaron 54 pacientes, donde se pudo
detectar duplicaciones internas en tándem (ITD) en 8/54
(14,8%) de los pacientes, de los cuales 5/8 (62,5%) de
ellos fueron positivos para la translocación MLL/AF4.
Dentro de este grupo, también se encontraron pacientes
positivos para la translocación ETV6/RUNX1 1/5 (20%);
translocación TCF3/PBX12/5 (40%) y pacientes con la
translocación BCR/ABL12/5 (40%), las cuales estuvieron
en conjunción con el transcrito MLL/AF4. Los otros 3/8
pacientes positivos ITD no presentaron ninguna de las
alteraciones moleculares aquí evaluadas. Asimismo,
5/8 (62,5%) pacientes ITD positivos fueron del sexo
masculino y 3/8 (37,5%) fueron del sexo femenino, en
donde la edad promedio fue de 10,8 años, presentándose
un adulto (12,5%) con una edad de 56 años y siete niños
(87,5%) con edades desde 0 hasta 10 años. Por otra parte
se destaca que, la mayoría de los pacientes positivos
para las mutaciones ITD/FLT3 presentaron un alto
porcentaje de blastos linfoides (datos no mostrados) en
comparación con los valores normales.
Ninguno de los pacientes analizados para la mutación
D835 en el exón 20 del gen FLT3 presentó el patrón
mutado, concluyendo así, que entre los pacientes
evaluados para esta mutación no hubo heterocigotos.
F) Alteraciones en el gen PAX5: 12/24 (50%) de las
muestras evaluadas para la mutación P80R, presentaron
un patrón de heterocigosis para dicha mutación. En el
análisis de secuencia de estas muestras se observó que
ninguna presentó la mutación P80R, sin embargo, dos de
las muestras evaluadas presentaron una sustitución de
la base C por T en la posición 686 de la hebra transcrita,
conduciendo a un codón TCT que sustituye al codón
CCT en el ARNm, el cual codifica al aminoácido Serina
en lugar de Prolina, dando origen a la mutación P80S.
Adicionalmente, uno de estos dos pacientes presentó
una deleción del triplete AGC el cual codifica para el
aminoácido serina en posición 77 de la proteína (Panel
A-Figura 2).
A su vez, se observó en uno de los pacientes positivos
para la mutación P80S, la presencia de dos mutaciones
silenciosas (la ubicación de estas mutaciones están
señaladas con flechas de color verde en el panel A de la
Figura 2). Así se tiene para este paciente, el triplete GGC
en lugar de GGA generando el aminoácido Glicina en
posición 76 de la proteína, y el triplete ATA en lugar de
ATT generando el aminoácido Isoleucina en posición 83
de la proteína en ambos casos. Por su parte, en el panel
B de la Figura 2 se pueden observar las diferencias en la
secuencia que presentan las muestras de los pacientes
en comparación con la proteína tipo silvestre (wt-wild
type) y con la proteína que se genera por la mutación
reportada en la bibliografía (mutación P80R).
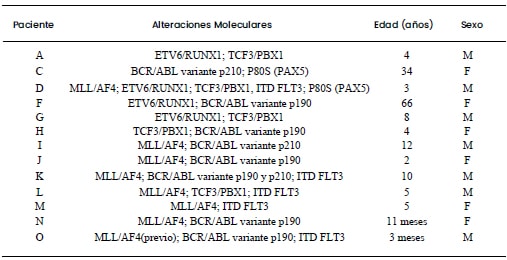
Es importante resaltar que en ambos pacientes (C y
D), además de la mutación puntual presente en el gen PAX5, también presentaron positividad para otras de las
alteraciones evaluadas (Tabla 3).
En otro orden de ideas, se evidenció mediante el uso
de la herramienta bioinformática SIFT, que la mutación
P80S es considerada perjudicial para el funcionamiento
de la proteína PAX5 (ENSP00000350844), asimismo,
el uso de PROVEAN permitió evidenciar que la deleción
S77del es considerada perjudicial para el funcionamiento
de la proteína PAX5.
Tabla 3. Coexistencia de alteraciones moleculares
DISCUSIÓN
La leucemia linfocítica aguda sigue siendo la principal
causa de muerte en niños y adultos jóvenes, aunque la
tasa de supervivencia se ubica en casi el 90% (Conter
et al., 2010; Pui et al., 2011). Las muestras biológicas
utilizadas para el diagnóstico de la LLA en la mayoría
de los casos, están compuestas por múltiples clones con
distintas alteraciones genéticas, lo cual puede influir
categóricamente en la respuesta a tratamiento, y a su
vez en el riesgo de reaparición del cuadro leucémico
(Mullighan et al., 2008).
Se ha determinado que tanto
en pacientes pediátricos como en pacientes adultos, el
proceso de transformación es usualmente iniciado por
una translocación cromosomal, creando un gen de fusión
con una actividad aberrante. Algunos de estos genes
de fusión pueden interferir en el nivel transcripcional
(MLL/AF4, ETV6/RUNX1, E2A/PBX1), mientras que otros
pueden activar rutas de señalización que promueven la
oncogénesis (BCR/ABL1) (Mullighan et al., 2008).
Algunos investigadores consideran que estas
alteraciones cromosómicas son un epifenómeno
del proceso oncogénico sin relación con su causa.
Sin embargo, este criterio cambió completamente
cuando, mediante las técnicas de biología molecular,
se puso en evidencia la existencia de genes específicos
comprometidos en las translocaciones cromosómicas
presentes en la mayor parte de las leucemias y que son
activados por la alteración (Pui et al., 2011).
En nuestro estudio se observó un predominio de
pacientes de sexo masculino tanto incluidos en el estudio,
como con presencia de alteraciones moleculares, lo que
es similar a lo reportado en la bibliografía (Pui et al.,
2015). Aunque el sexo y la etnia no son utilizados en la
estratificación de riesgo, tienen importancia pronóstico.
De hecho, se ha determinado que el sexo masculino
presenta peor pronóstico en comparación con el sexo
femenino (Hunger et al., 2012).
Son innumerables las investigaciones que han
permitido conocer el espectro de frecuencia con la que se
presenta esta patología en distintas partes del mundo, lo
que ha permitido a su vez ampliar tanto el conocimiento
que se tiene sobre la enfermedad, así como también
establecer comparaciones entre las distintas poblaciones
donde se ha reportado su incidencia. En el presente
estudio se realizó la determinación de las alteraciones
genéticas utilizando diferentes tipos de PCR en muestras
de 130 pacientes con diagnóstico clínico de leucemia
linfocítica aguda, en donde 56 de estos pacientes tuvieron
una o varias alteraciones genéticas. Las alteraciones
moleculares que identificamos en orden de frecuencia
fueron: t(9;22)(q34q11), t(12;21)(p13q22), t(11;19)
(q23p13.3) y t(4;11)(q21;q23) además de mutaciones
en el gen FLT3 y PAX5, en su mayoría relacionadas con
mal pronóstico. A pesar de nuestro pequeño tamaño
de muestras, los datos aquí obtenidos nos indican la
importancia que representa la identificación en forma
rutinaria de todas estas alteraciones.
Hasta el momento son pocas las publicaciones que
han reportado coexistencia de transcritos en pacientes
con LLA. La coexistencia de alteraciones se ha visto con
mayor frecuencia entre las translocaciones BCR/ABL
y TCF3/PBX1 (Jiménez Morales et al., 2008; Devaraj et
al., 1995). Sin embargo, los resultados obtenidos en el
presente estudio (Tabla 3) indican una alta frecuencia
de coexistencia de alteraciones genéticas en pacientes
venezolanos 13/130 (10%). La información aquí obtenida
aporta datos relevantes en cuanto a la heterogeneidad
clonal y la diversidad de cambios presentes en pacientes
LLA y a su vez aporta una idea en lo que respecta a
los efectos que cada una de estas alteraciones podría
ejercer a nivel de las células linfoides y a nivel de la
hematopoyesis en general.
La frecuencia del gen de fusión ETV6/RUNX1 en este
estudio (11,5%), estuvo por debajo de lo reportado en
gran parte de la literatura (25%) (Pui et al., 2008). Sin
embargo, trabajos realizados en México (7,2%) (Guerra
et al., 2016) y Tailandia (4,2%) (Limsuwanachot et al.,
2016), también han reportado porcentajes más bajo de
lo frecuente. Aunque la mayoría de las publicaciones
asocian esta alteración con un pronóstico favorable,
particularmente en la población pediátrica, existen
estudios que demuestran lo contrario; de hecho,
hay evidencia que la presencia de anormalidades
cromosómicas secundarias tales como la deleción del
alelo ETV6 no translocado, la duplicación del cromosoma
21, alteraciones genéticas submicroscópicas en los
genes EBF1, PAX5, BTLA, NR3C1, TOX, BMF, TBL1XR1 y
BTG1, influirían adversamente en el curso clínico de los
pacientes ETV6/RUNX1+ (Mullighan et al., 2008). Varios
de los genes antes mencionados están asociados con la
resistencia a drogas y a casos de recaída en pacientes con
LLA ETV6/RUNX1+ (Lindqvist et al., 2015).
Comparando la frecuencia obtenida en la población
venezolana para el oncogén BCR/ABL1 con lo observado en
otros países, podemos decir que tenemos una frecuencia
alta de presentación de este oncogén (específicamente
la variante p190 fue la que se presentó con mayor
porcentaje), particularmente tomando en cuenta
que la mayor parte de la población estudiada estuvo
conformada por pacientes pediátricos (Limsuwanachot
et al., 2016; Carranza et al., 2013). De manera interesante,
se lograron detectar pacientes que presentaron ambas
variantes al mismo tiempo (p190 y p210), todos
pertenecientes a la población pediátrica. Este último
hallazgo es de relevancia para nuestra población, ya que
la conjunción de ambas variantes ha sido observada con
mayor frecuencia en pacientes con leucemia mieloide
crónica (Ayatollahi et al., 2018), además de que nos
conduce a no restringir la identificación del gen de fusión
BCR/ABL1 en pacientes LLA, solo a la variante p190 por
ser la más común.
Por otra parte, el porcentaje de presentación de
mutación es ITD en el gen FLT3 en concordancia con
la presencia del encogen MLL/AF4 en varios de los
pacientes incluidos en este estudio, se correlacionan con
lo establecido en la bibliografía donde se ha indicado
la estrecha relación entre la expresión alterada del gen FLT3 y las alteraciones en MLL. También se ha observado
la actividad aumentada de FLT3 en conjunción con otros
genes de fusión t(12;21) ETV6/RUNX1 y t(9;22) BCR/ABL1
(Chillon et al., 2012) (Tabla 3). Otro estudio ha resaltado
que la asociación de mutaciones en FLT3 conjuntamente a
alteraciones en el gen MLL, se presentan en pacientes con
diagnóstico de leucemias bifenotípicas, las cuales cursan
con leucocitosis alto porcentaje de blastos en médula ósea
y mal pronóstico (Xu et al., 2005). La expresión alterada
de FLT3 puede predecir el pronóstico de las LLA t(4;11)+,
generando respuestas negativas, evidenciadas por una
supervivencia global y supervivencia libre de enfermedad
más corta. Este impacto de pronóstico negativo en los
pacientes LLA portadores de tales mutaciones en dicho
gen, viene dado a que la presencia de mutaciones (FLT3/
ITD) genera activación constitutiva del receptor FLT3 y puede desempeñar un papel en la supervivencia o la
proliferación de blastos leucémicos (Woo et al., 2014;
Kang et al., 2012). Lo anteriormente dicho sugiere que la
vigilancia constante del perfil de expresión de FLT3 en
pacientes LLA, podría conducir a un mejor seguimiento
en relación con el protocolo de terapia.
Montes et al. (2014) modelaron el efecto de FLT3 en
células de sangre de cordón humano (CD34) tranducidas
con MLL/AF4 y un FLT3 mutado. Observaron que, aunque
los ratones no desarrollaron leucemia, se presentó una
expansión transitoria de las células de la sangre del
cordón CD34 que expresaban MLL/AF4 tras la activación
de FLT3.
Por su parte, la mutación P80S encontrada en el
gen PAX5 se clasificó como una mutación con cambio
de sentido (missense mutation), cuya sustitución de
una única base genera un nuevo codón el cual codifica
un aminoácido diferente, generando alteración de la
estructura proteica, que viene dada por sustitución de
un aminoácido no polar por un aminoácido polar, que
puede establecer enlaces de hidrógeno con el agua,
atenuación de la capacidad de unión al ADN y de la
actividad de transactivación por la ubicación tanto de la
mutación P80S como de la deleción S77del en el dominio paired (este dominio puede funcionar como activador o
represor de la transcripción) de la proteína (Woo et al.,
2014).
Aunque las características clínicas, tales como el
tipo de leucemia, edad, alta carga tumoral, infiltración
al SNC, entre otras, han sido tradicionalmente usadas
para estimar el pronóstico de los pacientes, las
alteraciones genéticas específicas ofrecen una forma
más directa y quizá más segura para determinar la
progresión de la enfermedad. Por lo tanto, la detección
eficiente de fusiones génicas es de importancia crítica
para el diagnóstico, pronóstico y monitoreo después
del tratamiento. Además, se recomienda el uso de
metodologías más sensibles como la PCR en tiempo real,
análisis por microarreglos, identificación de SNPs (Pui et
al., 2015), tecnologías como la secuenciación de genoma
completo de siguiente generación (GWAS) y NGS, de
tal manera que se permita establecer un diagnóstico
certero y oportuno, además de contribuir al desarrollo
de fármacos dirigidos a inhibir la función de proteínas
oncogénicas y al diseño de esquemas de tratamiento
específicos que permitan mejorar la sobrevida libre de
enfermedad (SLE) y disminuir las probabilidades de
desarrollar efectos colaterales o resistencia a drogas
antineoplásicas. De tal forma que la leucemia debe ser
considerada y por ende, tratada como una enfermedad
compleja (Pui et al., 2015).
CONCLUSIONES
Mediante el uso de protocolos modernos de tratamiento en la LLA, se ha logrado mejorar hasta 80% la sobrevida libre de enfermedad, estos logros se deben principalmente al uso de terapias basadas en el riesgo, lo que implica dar un tratamiento con una intensidad específica para cada paciente en función del riesgo de recaída; de esta manera los pacientes no son subtratados ni sobretratados, lo cual disminuye los efectos colaterales y por lo tanto aumenta las probabilidades de curación. Sin embargo, en Venezuela la clasificación de riesgo de los pacientes con LLA con base a las características moleculares es aún limitado. Este trabajo presenta un panorama general de la frecuencia de las anormalidades genéticas más comunes descritas en otras poblaciones, y aunque hasta el momento no ha sido posible determinar el valor pronóstico de ellas en los pacientes venezolanos, estamos encontrando datos interesantes con respecto a la frecuencia de las alteraciones moleculares más relevantes en pacientes con LLA, lo cual plantea la urgente necesidad de realizar, tanto estudios multidisciplinarios en el que se aplique la combinación de herramientas citogenéticas y moleculares, como la necesidad de poner en práctica la aplicación de tecnologías moleculares más avanzadas de forma rutinaria, que permitan conocer las características genéticas de la LLA en los pacientes venezolanos, para desarrollar un algoritmo de diagnóstico que conduzca a una mejor clasificación genética de los pacientes por estratificación de riesgo, y por ende conlleven al desarrollo de mejores esquemas de tratamiento.
LIMITACIONES DEL ESTUDIO
La limitación de este estudio está relacionada con el diseño descriptivo y observacional. Sin embargo, es la base para iniciar un estudio en el que se busque la asociación de las alteraciones citogenéticas con las características clínico-biológicas de la enfermedad a largo plazo.
AGRADECIMIENTOS
Nos gustaría expresar nuestro agradecimiento al Ministerio de Ciencia y Tecnología por el otorgamiento de la Beca “Misión Ciencia” para estudios de postgrado y Fonacit Venezuela (proyecto G2001000784) por su apoyo financiero. A los Doctores y asistentes de los distintos centros hospitalarios por su colaboración y disposición en la obtención de muestras e historias clínicas de los pacientes, lo cual hizo posible la realización de esta investigación.
BIBLIOGRAFÍA
1. Abdullaev F., Rivera Luna R., Roitenburd Belacortu V., Espinosa Aguirre J. (2000) Pattern of childhood cancer mortality in Mexico. Arch. Med. Res. 31: 526-531.
2. Armstrong S.A., Kung A.L., Mabon M.E., Silverman L.B., Stam R.W., den Boer M.L. (2003) Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell. 3 (2): 173-183.
3. Armstrong S.A., Staunton J.E., Silverman L.B., Pieters R., den Boer M.L. (2002) MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30 (1): 41-7.
4. Ayatollahi H., Keramati M.R., Shirdel A., Kooshyar M.M., Raiszadeh M., Shakeri S. (2018) BCR-ABL fusion genes and laboratory findings in patients with chronic myeloid leukemia in northeast Iran. Caspian J. Intern. Med. 9 (1): 65-70.
5. Capote L. (2012) Perfil epidemiológico y control del cáncer en Venezuela. Academia Nacional de Medicina 4 (44).
6. Carranza C., Granados L., Morales O., Jo W., Villagran S., Tinti D., Villegas M., Antillón F., Torselli S., Silva G. (2013) Frequency of the ETV6-RUNX1, BCR-ABL1, TCF3-PBX1 and MLL-AFF1 fusion genes in Guatemalan pediatric acute lymphoblastic leukemia patients and their ethnic associations. Cancer Genet. 206 (6): 227-32.
7. Chillon M.C., Gomez Casares M.T., López Jorge C.E., Rodriguez Medina C., Molines A., Sarasquete M.E., Alcoceba M., Miguel J.D.G.S., Bueno C., Montes R., Ramos F., Rodríguez J.N., Giraldo P., Ramírez M., García Delgado R., Fuster J.L., González Díaz M., Menéndez P. (2012) Prognostic significance of FLT3 mutational status and expression levels in MLL-AF4+ and MLL-germlineacute lymphoblastic leukemia. Leukemia 26 (11): 2360-2366. doi:10.1038/leu.2012.161.
8. Chomczynski P., Sachi N. (1987). Single step method of RNA isolation by aid quanidiniumthiocyanate phenol-clhoroform extraction. Anal Biochem.162: 156-159.
9. Conter V., Aricò M., Basso G., Biondi A., Barisone E., Messina C. (2010) Long-term results of the Italian Association of Pediatric Hematology and Oncology (AIEOP) Studies 82, 87, 88, 91 and 95 for childhood acute lymphoblastic leukemia. Leukemia 24 (2): 255-64. doi: 10.1038/leu.2009.250.
10. Devaraj P.E., Foroni L., Kitra Roussos V., Secker Walker L.M. (1995) Detection of BCR-ABL and E2A-PBX1 fusion genes by RT-PCR in acute lymphoblastic leukaemia with failed or normal cytogenetics. Br. J. Haematol. 89 (2): 349-55.
11. Díaz Beveridge J., Urtasun Aparicio R. (2003) Leucemias agudas y síndromes mielodisplásicos secundarios al tratamiento oncológico. An. Med. Interna (Madrid) Vol. 20 (5): 257-268.
12. Globocan (2012) Cancer Incidence and Mortality Worldwide in 2012 http://www-dep.iarc.fr (referencia electrónica).
13. Guerra Castillo F.X., Ramos Cervantes M.T., Rosel Pech C., Jiménez Hernández E., Bekker Méndez V.C. (2016) PCR detection of relevant translocations in pediatric acute lymphoblastic leukemia. Rev. Med. Inst. Mex. Seguro Soc. 54 (3): S302-S308.
14. Hunger S.P., Lu X., Devidas M., Camitta B.M., Gaynon P.S., Winick N.J. (2012) Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J. Clin. Oncol. 30 (14): 1663-9.
15. Jiménez Morales S. (2002) Caracterización molecular de la leucemia aguda linfoblástica infantil. Tesis de Maestría. Universidad Autónoma Metropolitana, Iztapalapa, México.
16. Jiménez Morales S., Miranda Peralta E., Saldaña Álvarez Y., Pérez Vera P., Paredes Aguilera R., Rivera Luna R. (2008) BCR-ABL, ETV6- RUNX1 and E2A-PBX1: Prevalence of the most common acute lymphoblastic leukemia fusion genes in Mexican patients. Leuk. Res. 32 (10): 1518-22.
17. Kang H., Wilson C.S., Harvey R.C. (2012) Gene expression profiles predictive of outcome and age in infant acute lymphoblastic leukemia: a Children´s Oncology Group study. Blood 119: 1872-1881.
18. Landolfi C., Corredor M., Fernandez I., García R., González S., Latuff J., Rodríguez P., Tersek Y., Tovar E., Urdaneta B., Acosta M., Araujo C., Catalayud J., Collazo M., Costa M., Cova J., DiStefano M., Díaz M., Duerto M., García R., González L., López J., Lugo I., Mejía M., Pachano S., Ramírez F., Rojas V., Salazar E., Salazar M., Sánchez M., Sánchez P., Vizcaíno J., Zavahra M., Chacín M., Gross A., Oliveros A., Prado A., Travieso B., Aponte B., Castro Y., Cedres S., Deninzon L., Mendoza F., Ramírez R., Soto A. (2013) I Consenso Venezolano sobre leucemia aguda de la infancia y adolescencia. Sociedad Venezolana de Hematología.
19. Limsuwanachot N., Siriboonpiputtana T., Karntisawiwat K., Chareonsirisuthigul T., Chuncharunee S., Rerkamnuaychoke B. (2016) Multiplex RT-PCR Assay for Detection of Common Fusion Transcripts in Acute Lymphoblastic Leukemia and Chronic Myeloid Leukemia Cases. Asian Pac. J. Cancer Prev. 17 (2): 677-84.
20. Lindqvist C.M., Nordlund J., Ekman D., Johansson A., Moghadam B.T., Raine A. (2015) The mutational landscape in pediatric acute lymphoblastic leukemia deciphered by whole genome sequencing. Hum. Mutat. 36: 118-128.
21. Montes R., Ayllon V., Prieto C., Bursen A., Prelle C., Romero Moya D. (2014) Ligandindependent FLT3 activation does not cooperate with MLL-AF4 to immortalize/ transform cord blood CD34+ cells. Leukemia 28 (3): 666-674. doi:10.1038/leu.2013.346.
22. Mullighan C.G., Phillips L.A., Su X., Ma J., Miller C.B., Shurtleff S.A. (2008) Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 322 (5906): 1377-80. doi: 10.1126/science.1164266.
23. Pulte D., Gondos A., Brenner H. (2009) Improvement in survival in younger patients with acute lymphoblastic leukemia from the 1980s to the early 21st Century. Blood 113 (7): 1408-11.
24. Pui C.H., Carroll W.L., Meshinchi S., Arceci R.J. (2011) Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J. Clin. Oncol. 29 (5): 551-65.
25. Pui C.H., Robison L.L., Look A.T. (2008) Acute lymphoblastic leukaemia. Lancet 371: 1030- 43.
26. Pui C.H., Yang J.J., Hunger S.P., Pieters R., Schrappe M., Biondi A. (2015) Childhood Acute Lymphoblastic Leukemia: Progress Through Collaboration. J. Clin. Oncol. 233 (27): 2938-48.
27. Smith M.A., Ries L.A., Gurney J.G. (1999) Leukemia. In: Ries L.A., Smith M.A., Gurney J.G. (Eds.) Cancer incidence and survival among children and adolescents: United States SEER Program 1975-1995. Bethesda, Md: National Cancer Institute, SEER Program, 1999. NIH Pub. N° 99-4649., pp. 17-34. Also available online. Last accessed October 19, 2010.
28. Stam R.W., den Boer M.L., Schneider P., Nollau P., Horstmann M., Beverloo H.B. (2005) Targeting FLT3 in primary MLL-generearranged infant acute lymphoblastic leukemia. Blood 106 (7): 2484-90.
29. Teitell M.A., Pandolfi P.P. (2009) Molecular genetics of acute lymphoblastic leukemia. Annu. Rev. Pathol. 4: 175-98.
30. Tirado L., Mohar A. (2007) Epidemiología de las Neoplasias Hematológicas. Rev. Inst. Nal. Cancerol. 2: 109-120.
31. Villalta D., Sajo Castelli A.M, Ovalles P. (2019) Pronósticos de la mortalidad e incidencia de cáncer en Venezuela año 2019. Sociedad Anticancerosa de Venezuela.
32. Wiemels J. (2012) Perspectives on the causes of childhood leukemia. Chem. Biol. Interact. 196: 59-67.
33. Woo J., Alberti M., Tirado C. (2014) Childhood B-acute lymphoblastic leukemia: a genetic update. Experimental Hematology & Oncology 3: 16.
34. Xu B., Li L., Tang J.H., Zhou S.Y. (2005) Detection of FLT3 gene and FLT3/ITD mutation by polymerase chain reaction-single-strand conformation polymorphism in patients with acute lymphoblastic leukemia. Di Yi Jun Yi Da XueXueBao 25 (10): 1207-10.