


Vol. XXXI Issue 2
Article 5
DOI: 10.35407/bag.2020.31.02.05
ARTÍCULOS
ORIGINALES
Alelos
asociados al índice de severidad de la enfermedad Mal de Río Cuarto en
germoplasma exótico de maíz
Alleles associated to
disease severity index of Mal de Río Cuarto disease in maize exotic germplasm
Rossi E.A.1,2
*
Ruiz M.1,2
Bonamico N.C.1,2
Balzarini M.G.3,4
1 INIAB-Instituto de Investigaciones Agrobiotecnológicas (CONICET-UNRC).
2 Facultad de Agronomía y Veterinaria, Universidad Nacional
de Río Cuarto.
3 CONICET-Consejo Nacional de Investigaciones Científicas y
Técnicas.
4 Facultad de Ciencias Agropecuarias, Universidad Nacional de
Córdoba.
* Corresponding author: Ezequiel Rossi erossi@ayv.unrc.edu.ar
ORCID 0000-0003-2043-6760
RESUMEN
El Mal de Río Cuarto (MRC) es una de las
enfermedades virales más importantes del maíz en Argentina. El índice de
severidad de enfermedad (ISE) permite combinar la incidencia y la severidad de
una enfermedad en una métrica única. La reacción genotípica a MRC ha sido muy
estudiada en poblaciones biparentales, sin embargo este carácter complejo no se
ha analizado mediante estudios de mapeo por asociación. El objetivo del
presente trabajo es identificar nuevos alelos de resistencia asociados con el
ISE de la enfermedad MRC de maíz en un germoplasma exótico del Centro
Internacional de Mejoramiento de Maíz y Trigo (CIMMYT). Una población de líneas
de maíz del CIMMYT se evaluó fenotípicamente en ambientes donde la enfermedad
es endémica. Los predictores del efecto genotípico (BLUP, best linear
unbiased predictor) del ISE de MRC y 78.376 marcadores SNP (Single
Nucleotide Polymorphism) se usaron para realizar el mapeo por asociación en
186 líneas de maíz. Los componentes de varianza y los valores de heredabilidad
sugieren una amplia variabilidad genotípica en la población de líneas. El mapeo
por asociación permitió identificar 11 posibles QTL de resistencia a MRC. La
incorporación de germoplasma exótico en los programas de mejoramiento de maíz
locales podría contribuir favorablemente a la creación de genotipos híbridos
con mayor nivel de resistencia a MRC. La capacidad predictiva de los marcadores
asociados con la resistencia a MRC indican que la selección asistida por
marcadores es una herramienta recomendable para seleccionar genotipos
resistentes a MRC.
Palabras clave: Índice de severidad de
enfermedad; Mapeo por asociación; QTL; SNP
ABSTRACT
Mal de Río Cuarto (MRC) is one of the most important viral diseases of
maize in Argentina. The disease severity index (DSI) allows to combine the
incidence and severity of a disease in a single metric. The genotypic reaction
to MRC has been extensively studied in biparental populations. However, this
complex trait has not been analyzed by genome-wide association studies (GWAS).
The aim of this work is to identify new resistance alleles associated with DSI
of MRC in an exotic germplasm from the International Maize and Wheat
Improvement Center (CIMMYT). A population of maize lines from CIMMYT was
phenotypically evaluated in environments in the area where the disease is
endemic. The predictors of genetic effects (BLUP, best linear unbiased
predictor) and 78,376 SNP markers (Single Nucleotide Polymorphism) were used to
perform the GWAS in 186 maize lines. The values of variance components and
mean-basis heritability suggest a wide genotypic variability in the population.
The GWAS allowed to identify 11 putative QTL of resistance to MRC. The
incorporation of exotic germplasm into local maize breeding programs could
contribute favorably to the creation of hybrids with a higher level of
resistance to MRC. The predictive ability of associated markers with MRC
resistance indicates that marker-assisted selection is an advisable tool for
selecting MRC resistant genotypes.
Key words: Disease severity index; Genome-wide
association study; QTL; SNP
Received: 05/12/2020
Revised version received: 09/15/2020
Accepted: 10/14/2020
INTRODUCCIÓN
El maíz (Zea mays L.) es
hospedante natural de más de 50 virus (Lapierre y Signoret, 2004), y una de las enfermedades virales más
importantes en Argentina es el Mal de Río Cuarto (MRC) (Gimenez Pecci et al., 2012). El agente causal de la enfermedad MRC es el Mal
de Río Cuarto virus (MRCV) perteneciente a la familia Reoviridae, género Fijivirus
(Distéfano
et al., 2002). Este
virus es naturalmente transmitido en forma persistente, cíclica y propagativa
por medio de insectos, siendo la chicharrita Delphacodes kuscheli Fennah
el principal vector de la enfermedad (Remes Lenicov et al., 1985). La forma de transmisión indica que el vector
es reservorio natural del virus y la población de macrópteros migrantes
constituye el principal inóculo de la enfermedad (Ornaghi et al., 1993). El principal síntoma de la enfermedad MRC es
la presencia de enaciones o protuberancias sobre las nervaduras en el envés de
las hojas. Las plantas pueden presentar otros síntomas como resultado de las
modificaciones en los niveles hormonales endógenos, tales como tallos
achatados, entrenudos cortos, hojas del tercio superior recortadas o reducidas
a la vaina foliar, panojas atrofiadas de tamaño reducido y espigas múltiples
con pocos o sin granos (Giménez Pecci et al., 2012). Las siembras tempranas
y el uso de agroquímicos son estrategias de manejo agronómico para reducir la
enfermedad MRC mediante el control del insecto vector, pero la siembra de
genotipos resistentes es el proceder recomendado (Di Renzo et al., 2004). La reacción a la enfermedad MRC se comporta
como un carácter cuantitativo (Di Renzo et al., 2002). Estudios
moleculares de la reacción genotípica a MRC, han permitido identificar alelos
de resistencia en poblaciones biparentales (Di Renzo et al., 2004; Bonamico et al., 2012; Rossi et al., 2015). Sin embargo, en este tipo de mapeo pueden
perderse alelos que no segregan entre los parentales de la población y alelos
asociados a loci de efecto menor (Warburton et al., 2015). La creciente disponibilidad de datos
genómicos polimórficos y el potencial para explorar múltiples eventos de
recombinación ocurridos en la historia evolutiva de un germoplasma específico,
han convertido a los estudios de mapeo por asociación (GWAS, del inglés genome
wide association studies) en una importante alternativa para estudiar
caracteres de variación cuantitativa en plantas (Hao et al., 2015). La respuesta de los genotipos a enfermedades
virales es comúnmente estimada mediante los caracteres incidencia y severidad
(Rossi et al., 2019b). Sin embargo, en MRC existen diversos síntomas que
se manifiestan en distintos grados de severidad y con distinta incidencia en
las poblaciones evaluadas. Para contemplar esta característica de la enfermedad
se han realizado estudios donde los caracteres incidencia y severidad se han
sintetizado o resumido en un índice, como es el caso del índice de severidad de
enfermedad (ISE) (Shi et al., 2012; Bonamico et al., 2013; Chen et al., 2015). Este índice constituye una media de la
severidad de la reacción al MRC, ponderada por la incidencia de los distintos
grados de severidad observada en un conjunto de plantas de cada genotipo. Si
bien se han realizado análisis de QTL en poblaciones biparentales para el ISE
de MRC en la zona de Argentina donde la enfermedad es endémica (Bonamico et
al., 2012; Di Renzo et al., 2004), no se ha analizado este carácter
en el contexto de GWAS, principalmente por la falta del fenotipado en
poblaciones de gran tamaño extensamente genotipadas. El Centro Internacional de
Mejoramiento de Maíz y Trigo (CIMMYT) es una importante fuente de germoplasma
para la incorporación de alelos exóticos en los programas de mejoramiento de
todo el mundo. Las líneas de maíz de CIMMYT han sido extensivamente genotipadas
mediante marcadores SNPs (Wu et al., 2016) y son adecuadas para realizar estudios de
mapeo por asociación en distintas partes del mundo donde puedan ser evaluadas
fenotípicamente; algunas de estas líneas ya han mostrado buena adaptación en la
zona de Argentina donde la enfermedad es endémica (Rossi et al., 2019a).
El objetivo del presente trabajo fue identificar nuevos alelos de resistencia asociados
al ISE de la enfermedad MRC de maíz en un germoplasma exótico de CIMMYT.
MATERIALES Y MÉTODOS
Material vegetal y ensayo de campo
Una población de 210 líneas endocriadas
de maíz proveniente del CIMMYT se evaluó fenotípicamente para la reacción a la
enfermedad viral Mal de Río Cuarto en la zona donde la enfermedad es endémica.
La población fue caracterizada por su adaptación a la región sur de Córdoba y
su diversidad genotípica por Rossi et al. (2019a). Los ensayos de campo se realizaron en las
localidades de Río Cuarto (64° 20′ W, 33° 8′ S) y Sampacho (64º
44′ W, 33º 20′ S), Córdoba, Argentina, bajo condiciones de
transmisión natural. Los ensayos se establecieron en los ciclos agrícolas
2018-2019 y 2019-2020 en Río Cuarto y en el ciclo agrícola 2018- 2019 en
Sampacho. Cada combinación año-localidad se consideró un ambiente de análisis:
Río Cuarto 2018- 2019 (RC-18-19), Río Cuarto 2019-2020 (RC-19-20) y Sampacho
2018-2019 (SA-18-19). Cada genotipo se sembró en parcelas de un surco de 2,5 m
de largo. En cada ambiente se utilizó un diseño parcialmente repetido (Cullis et al., 2006) donde un 25% de los genotipos se repitió tres
veces y el 75% de los genotipos restantes se sembró en una sola parcela, sin
repeticiones. El 25% de los genotipos repetidos fue diferente en cada ambiente
de acuerdo a la disponibilidad de semillas. Las plantas de cada parcela se
evaluaron individualmente según el grado de severidad propuesto por Ornaghi et al. (1993): 0=planta sin síntomas; 1=presencia de
enaciones; 2=presencia de enaciones + acortamiento de entrenudos + láminas
foliares atrofiadas en el tercio superior; 3=máximo desarrollo de la enfermedad
con enaciones + acortamiento de entrenudos + láminas foliares atrofiadas en el
tercio superior + espigas pequeñas, múltiples y sin granos. Para cada parcela
se estimó el índice de severidad de enfermedad (ISE) según Di Renzo et al. (2002), el cual involucra los caracteres incidencia
y severidad de MRC. Este índice puede tomar valores de 0 a 100, donde 0
corresponde a genotipos sin síntomas y 100 a genotipos severamente afectados.

Datos genotípicos
La caracterización molecular de la
colección de líneas de maíz de CIMMYT fue realizada por Wu et al. (2016). Un total de 362.008 marcadores SNP,
obtenidos mediante genotipado por secuenciación, está disponible públicamente
en el repositorio de datos de CIMMYT (http://data.cimmyt.org/dvn). A partir del
total de marcadores, se seleccionaron 78.376 SNP con una tasa de datos
faltantes menor al 35% y una mínima frecuencia alélica superior a 0,05.
Análisis estadístico
Los datos fenotípicos multi-ambientales
se analizaron con la función “mmer” del paquete “sommer” (Covarrubias Pazaran, 2016) en el software R (R Core Team 2016). Para
obtener la mejor predicción lineal insesgada (BLUP) (West et al., 2007) de los efectos de genotipos a través de
ambientes, se ajustó un modelo lineal mixto (MLM) multi-ambiental. El modelo
lineal mixto ajustado incluyó efectos de ambientes fijos, efectos de genotipos
aleatorios y efectos de interacción genotipo-ambiente (GE) aleatorios:
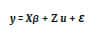
donde y es el vector de las
observaciones fenotípicas, X y Z son las matrices
de diseño. El vector β representa los efectos ambientales
considerados como fijos, u representa los efectos aleatorios de
genotipo y de la interacción GE y ε representa el error
aleatorio. Los efectos genéticos aleatorios se asumieron normalmente
distribuidos N (0, ), con la matriz de varianzacovarianza (G) con
estructura diagonal. Los efectos de la interacción GE se asumieron normalmente
distribuidos con media cero y matriz de varianza-covarianza no estructurada.
Los errores se asumieron normalmente distribuidos i.i.d. N (0, ) e
independientes del resto de efectos aleatorios. Ajustado el modelo se obtuvo el
mejor predictor lineal insesgado (BLUP) del efecto de genotipo. El valor del
BLUP para cada genotipo mide el efecto genético de la línea, descontando el
efecto ambiental y la potencial interacción GE, y este valor fue usado como
variable respuesta en el estudio de mapeo por asociación con el perfil
molecular. La heredabilidad del ISE, se estimó a partir de los componentes de
varianza obtenidos del MLM ajustado de acuerdo a Holland et al. (2010).
En ambientes individuales:
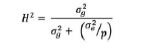
A través de ambientes:
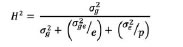
donde es el componente de varianza
genotípica, es el componente de varianza de la interacción GE, es la varianza
residual, e es el número de ambientes, y p es la media ponderada
del número de repeticiones de cada genotipo a través de ambientes.
Mapeo por asociación
El software TASSEL 5.2.60 (Bradbury et al., 2007) se usó para realizar la asociación entre los
78.376 SNPs y los BLUPs del ISE a través de ambientes para la población de
líneas de maíz con información fenotípica y genotípica (n=186). Se compararon
distintos modelos estadísticos de asociación (GWAS) para establecer el de mejor
ajuste a los datos del estudio: 1) modelo lineal general propuesto por Pritchard et al. (2000), con la estructura poblacional determinada
por la matriz Q con tres grupos (Wu et al., 2016), como covariable de efecto fijo (modelo Q);
2) modelo lineal general propuesto por Price et al. (2006), con la estructura poblacional determinada
mediante análisis de componentes principales, como covariable de efecto fijo
(modelo PCA); 3) modelo lineal mixto propuesto por Parisseaux y Bernardo (2004), con la matriz de parentesco relativo entre
individuos (Kinship) como efecto aleatorio (modelo K); 4) modelo lineal
mixto con la matriz kinship como efecto aleatorio y la matriz Q como
covariable de efecto fijo (modelo Q+K) (Yu et al., 2006); 5) modelo lineal mixto con la matriz kinship
como efecto aleatorio y las componentes principales como covariable de
efecto fijo (modelo PCA+K) (Zhao et al., 2007). La comparación entre modelos se realizó
mediante el gráfico de distribución empírica (Q-Q plot) que permite
evaluar la desviación de los valores de probabilidad (-log10 de valores-P)
observados respecto a los esperados bajo la hipótesis de asociación nula entre
el marcador y la variable respuesta, i.e. el valor BLUP de la línea para
el carácter ISE. Se utilizó el procedimiento propuesto por Li y Ji (2005) para corregir los valores-P y evitar
el incremento de la tasa de falsos positivos debido a la multiplicidad de
pruebas de hipótesis que se realizan al evaluar la asociación marcador por
marcador. La significancia estadística, para declarar asociación significativa
entre un marcador y el ISE, se definió en el valor umbral de -log10 (valor-P)
>4 (valor P<0.0001). El gráfico de distribución empírica (Q-Q
plot) y el gráfico Manhattan plot, donde se muestran los resultados
de las pruebas de asociación marcador por marcador, se realizaron con el
paquete “qqman” (Turner, 2018) del software R (R Core Team 2016) usando el
modelo de asociación seleccionado. Para predecir el efecto genotípico del ISE
de MRC se ajustaron dos modelos de predicción genómica usando los méritos
genéticos de las líneas como variable respuesta (i.e. BLUP de los
efectos de genotipo del ISE). En el primer modelo, se consideraron como
predictores todos los marcadores SNP utilizados en el mapeo por asociación. En
el segundo modelo, solo se consideraron los marcadores SNP que resultaron
estadísticamente asociados en el GWAS. El modelo de predicción genómica utilizado
en ambos casos fue el modelo Bayes C propuesto por Habbier et al.
(2011). La implementación de este modelo se realizó mediante el paquete BGLR
(Pérez Rodríguez y de los Campos, 2014) del software R (R Core Team 2016). La
eficiencia de la predicción se evaluó mediante validación cruzada. En cada
repetición de la validación, los datos se dividieron aleatoriamente en un
conjunto de entrenamiento (70% de las observaciones) y el conjunto
complementario se usó para validar las predicciones del mérito genético, realizada
a partir de uno u otro conjunto de marcadores (30% de las observaciones). La
validación se repitió 50 veces con asignaciones aleatorias independientes de
las líneas a los conjuntos de datos de entrenamiento y validación. La
eficiencia de la predicción, con uno u otro conjunto de marcadores, se
cuantificó utilizando la correlación entre los méritos genéticos de cada línea
y los méritos predichos por el modelo de predicción genómica. La correlación se
calculó en las 50 repeticiones aleatorias del proceso.
RESULTADOS
El ISE de MRC de las líneas de maíz, al
considerar los BLUPs a través de ambientes, sugiere una amplia variabilidad
genotípica en el grupo de líneas evaluadas (Figura 1). La Tabla 1 muestra los valores medios del
ISE en los tres ambientes de evaluación y a través de ambientes. Los tres
ambientes de evaluación presentaron un adecuado nivel de transmisión natural
del virus. El ensayo realizado en la localidad de Sampacho en el ciclo agrícola
2018-2019 (SA-18-19) fue el que presentó el mayor valor medio para el ISE. Los
componentes de varianza genotípica fueron altamente significativos en los tres
ambientes individuales y a través de ambientes. El componente de varianza de la
interacción genotipo × ambiente también fue significativo pero pequeño respecto
de la varianza genotípica a través de ambientes (Tabla 1). La heredabilidad
osciló entre 0,46 y 0,60 en ambientes individuales y fue de 0,70 a través de
ambientes.
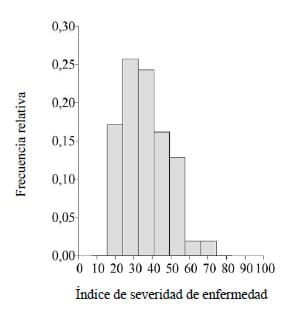
Figura 1. Frecuencia de distribución relativa para el
índice de severidad de enfermedad (ISE) de Mal de Río Cuarto a través de
ambientes.
Tabla 1. Medias, mínimo, máximo, componentes de varianza
y heredabilidad (H2) del índice de severidad de enfermedad (ISE) de Mal
de Río Cuarto (MRC) estimados a partir de 210 líneas de maíz en ambientes individuales
y a través de ambientes de la zona donde la enfermedad es endémica.
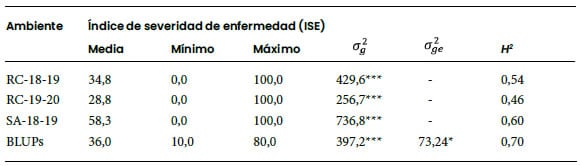
RC-18-19: Río Cuarto 2018-2019; RC-19-20: Río
Cuarto 2019-2020; SA-18-19:
Sampacho 2018-2019. componente de
varianza genotípica; componente de varianza de la interacción genotipoambiente.
*** <0,0001; *<0,05. Rango de ISE: 0-100%.
Los gráficos de distribución empírica (Q-Q
plot) permitieron comparar entre los distintos modelos de mapeo por asociación
utilizados. El modelo Q+K presentó una distribución sesgada hacia los valores
significativos. Los modelos K, PCA y PCA+K mostraron algunos puntos por debajo
de la diagonal, indicando una posible sobre-parametrización (datos no
mostrados). El modelo Q, que considera una estructura genética de la población
de tres grupos, fue el que mejor ajustó para realizar la asociación entre los
SNP y los BLUP del ISE (Figura 2). Un total de 14 SNP resultaron
estadísticamente asociados con el ISE a través de ambientes con un valor umbral
de -log10 valor P>4,00 (valor P<0,0001) (Figura 2, Tabla 2). Según la posición física de los
SNP, estos se agruparon en 11 posibles QTL de resistencia a MRC ubicados en los
cromosomas 1, 2, 4, 6, 7, 8 y 9. El porcentaje de variación fenotípica
explicada en forma individual por cada QTL osciló entre 10 y 16% (Tabla 1). Los
efectos del QTL 2 (bin 1.04) y del QTL 6 (bin 2.09) resultaron estadísticamente
no significativos. Los QTL 9, 4 y 11, ubicados en los bin 7.01, 2.02 y 9.04
respectivamente, fueron los que presentaron mayor efecto genético aditivo (Tabla
2). La eficiencia de la predicción fue en promedio de r=0,30 para el modelo
donde se incluyeron todos los SNPs y aumentó a r=0,60 cuando sólo se incluyeron
en el modelo el conjunto reducido de SNPs conformado con aquellos marcadores
estadísticamente asociados al ISE de MRC según los resultados del GWAS (Figura 3).
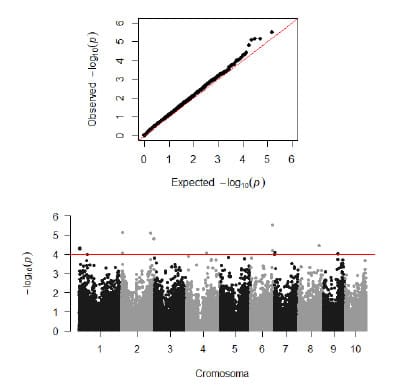
Figura 2. Distribución empírica (Q-Q plot) (arriba)
y pruebas de asociación marcador por marcador (Manhattan plot) (abajo),
de los resultados del mapeo por asociación para el índice de severidad de la
enfermedad (ISE) de Mal de Río Cuarto a través de ambientes.
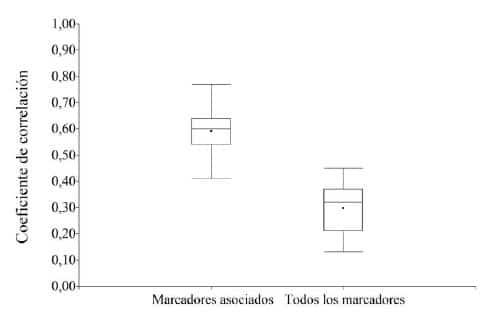
Figura 3. Distribución de la correlación entre los
valores observados de los BLUP del índice de severidad de la enfermedad (ISE)
Mal de Río Cuarto y los valores predichos por el modelo de predicción
construido a partir de los marcadores SNPs identificados como
significativamente asociados en el mapeo y a partir de todo el conjunto de SNPs
disponibles.
Tabla 2. Detalles de los marcadores SNP asociados con el
índice de severidad de enfermedad (ISE) de Mal de Río Cuarto.
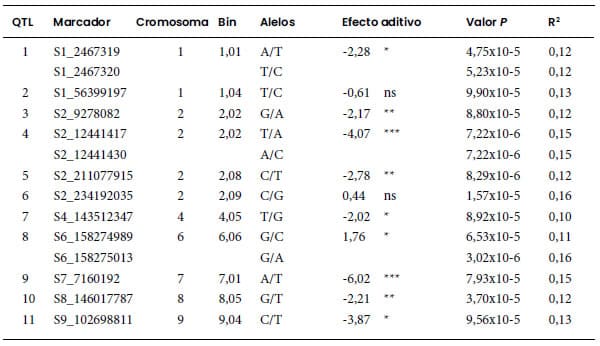
R2: variación fenotípica explicada por cada
marcador. La posición física exacta de cada SNP se puede inferir a partir del
nombre del marcador, por ejemplo, S9_102698811: cromosoma 9; 102.698.811 pb. El
alelo subrayado indica el alelo de resistencia para cada marcador.
DISCUSIÓN
La evaluación fenotípica precisa y
confiable es uno de los principales requisitos para realizar estudios de mapeo
por asociación (GWAS) (Yu et al., 2008). En maíz, la enfermedad viral MRC es
transmitida solamente por insectos, siendo el principal vector la chicharrita Delphacodes
kuscheli, por lo que la evaluación fenotípica se debe realizar bajo
condiciones de transmisión natural de la enfermedad. La variación de los
valores extremos del ISE de MRC es común entre ambientes de evaluación. En el
presente estudio, todos los ambientes registraron valores extremos del ISE y
por lo tanto permitieron diferenciar a los genotipos. De todos modos, para
evitar posibles efectos ambientales, se decidió utilizar los BLUP a través de
ambientes como variable indicadora de la performance de cada línea
durante el análisis de asociación con el perfil molecular. Los componentes de
varianza genotípica y los valores de heredabilidad estimados en el presente
estudio indican una amplia variabilidad genotípica en la población de líneas de
maíz proveniente del CIMMYT. Los valores de heredabilidad del ISE, estimados en
ambientes individuales y a través de ambientes, son superiores a los reportados
por Di Renzo et
al. (2002),
quienes evaluaron una población de familias F2:3 en tres ambientes de la zona
donde la enfermedad MRC es endémica. En estudios de mapeo por asociación, la
elección del modelo a utilizar es un paso importante para considerar los
efectos que pueden tener la estructura poblacional presente y las relaciones de
parentesco entre los genotipos en las asociaciones (Gutiérrez et al.,
2011). En nuestro trabajo, el modelo que mejor ajustó fue el que considera la
estructura poblacional determinada por la matriz Q como covariable de efecto
fijo. En este caso la matriz Q incluida se compone de tres grupos de acuerdo a
la adaptación ambiental de las líneas de maíz. Esta estructura de la población
fue propuesta por Wu et al. (2016), quienes caracterizaron molecularmente la
colección completa de 538 líneas de maíz de CIMMYT que incluye a la población
evaluada en el presente estudio. La integración de datos fenotípicos y
genotípicos podría aumentar potencialmente la eficiencia del mejoramiento de
caracteres complejos (Guo et al., 2020) como la resistencia a MRC. Si bien las bases
genéticas de la enfermedad MRC han sido ampliamente estudiadas en poblaciones
biparentales (Di Renzo et al., 2004; Bonamico et al., 2012), la evaluación de poblaciones diversas de
germoplasma exótico ha sido muy poco explorada. En el presente estudio, el
mapeo por asociación permitió identificar 11 posibles QTL para resistencia a
MRC. Al comparar los QTL identificados para el ISE con los identificados en
estudios previos, tanto para el ISE como para los caracteres incidencia y
severidad, se observan coincidencias en las posiciones físicas y en la
proporción de la variación fenotípica explicada. Por ejemplo, en el bin 4.05,
donde nosotros identificamos un SNP asociado al ISE, Bonamico et al.
(2013) identificaron un marcador microsatélite (SSR) para el ISE de MRC, al
evaluar una población de líneas endocriadas recombinantes (RILs). En el bin
1.01, Bonamico et al. (2012) identificaron un marcador SSR asociado con
incidencia y con severidad de MRC. En el mismo bin, en nuestro trabajo
identificamos un SNP asociado al ISE de la enfermedad. Redinbaugh et al. (2018) combinaron resultados de estudios donde se
identificaban QTL para resistencia a enfermedades virales en maíz e
identificaron nueve regiones del genoma donde se agrupaban loci de resistencia
(clusters). Uno de esos clusters se ubica en el bin 2.08 y
reporta QTL de resistencia para tres enfermedades virales. En el mismo bin y en
una posición física (pares de bases) muy próxima, identificamos un marcador
asociado con el ISE de MRC. Los QTL identificados en este estudio, en los bin
7.01 y 9.04 podrían considerarse como nuevos alelos de resistencia aportados
por el germoplasma exótico de CIMMYT debido a que en estudios previos no han
sido reportados QTL para MRC en esas posiciones. La relación entre incidencia y
severidad de enfermedades vegetales es epidemiológicamente importante (Seem, 1984). Esta relación puede ser estudiada según la
correlación genética entre ambos caracteres como se realizó en el trabajo de Rossi et al. (2020). Otra manera de estudiar la relación es a
través de la estimación del ISE, el cual combina la incidencia y la severidad
de una enfermedad en un único índice. Esto explica que los posibles QTL 1, 4, 8
y 9, identificados en este estudio, fueron también identificados por Rossi et
al. (2020) al evaluar la reacción a MRC mediante los caracteres incidencia
y severidad individualmente. Según Dintinger et al.
(2005) los QTL asociados a incidencia y severidad
podrían involucrar mecanismos de resistencia que reducen la evolución de
síntomas severos en la planta, y mecanismos de resistencia que reducen la
probabilidad que una planta manifieste síntomas. La validación cruzada del
modelo de predicción genómica demostró la capacidad predictiva de los
marcadores asociados al ISE de MRC, identificados en el GWAS. La menor
capacidad predictiva del modelo que incluye todos los marcadores podría estar
relacionada a correlaciones entre los marcadores y de éstos con el efecto
ambiente, los cuales podrían enmascarar el efecto de otros marcadores. Si bien
se requieren numerosos estudios para avalar el desempeño de la predicción
genómica para la resistencia a MRC en maíz, los resultados indican que la
selección asistida por marcadores basada en los SNPs que se encontraron
asociados al ISE de MRC podría ser una herramienta muy útil para la selección.
El uso del genotipado mediante PCR específica de alelos (KASP, Kompetitive
Allele Specific PCR), técnica de laboratorio que permite genotipar alelos
específicos de unos pocos marcadores, combinado con selección asistida por
marcadores, representa una alternativa más económica y rápida que la selección
genómica. Los resultados sugieren que el germoplasma exótico de CIMMYT tiene
amplia variabilidad genética para la resistencia a la enfermedad MRC. La
evaluación de un germoplasma exótico para la reacción a una enfermedad endémica
local contribuye a validar regiones genómicas identificadas en estudios previos
e identificar nuevos alelos de resistencia. En la industria semillera
argentina, la importación de paquetes tecnológicos “cerrados” es difícil porque
se requiere de adaptación a las condiciones agroecológicas de cada región del
país. Por lo tanto, la incorporación de germoplasma exótico adaptado a las
condiciones agroecológicas locales, podría favorecer la creación de genotipos
híbridos con mayor nivel de resistencia a MRC en los programas de mejoramiento
de maíz. La eficiencia predictiva de los marcadores asociados con la
resistencia a MRC indican que la selección asistida por marcadores constituye
una herramienta promisoria para seleccionar genotipos resistentes a MRC.
BIBLIOGRAFÍA
Bonamico N.C., Di Renzo M.A., Borghi M.L., Ibañez M.A., Díaz D.G., Salerno
J.C., Balzarini M.G. (2013) Mapeo de QTL
para una medida multivariada de la reacción al virus del Mal de Río Cuarto. J. Basic Appl. Genet. 24:
11-21.
Bonamico N.C., Di Renzo M.A., Ibañez M.A., Borghi M.L., Díaz D.G., Salerno
J.C., Balzarini M.G. (2012) QTL analysis of resistance to Mal de Río Cuarto
disease in maize using recombinant inbred lines. J. Agric. Sci. 150: 619-629. https://doi.org/10.1017/S0021859611000943.
Bradbury P.J., Zhang Z., Kroon D.E., Casstevens T.M., Ramdoss Y., Buckler
E.S. (2007) TASSEL: Software for association mapping of complex traits in
diverse samples. Bioinformatics. https://doi.org/10.1093/ bioinformatics/btm308.
Chen G., Wang X., Hao J., Yan J., Ding J. (2015) Genome-wide association
implicates candidate genes conferring resistance to maize rough dwarf disease
in maize. PLoS ONE 10: 1-13. https://doi.org/10.1371/journal.pone.0142001.
Covarrubias Pazaran G. (2016) Genome-Assisted prediction of quantitative
traits using the r package sommer. PLoS ONE 11: 1-15.
https://doi.org/10.1371/journal.pone.0156744.
Cullis B.R., Smith A.B., Coombes N.E. (2006) On the design of early
generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 11:
381-393. https://doi.org/10.1198/108571106X154443.
Dintinger J., Verger D., Caiveau S., Risterucci A.M., Gilles J., Chiroleu
F., Hamon P. (2005) Genetic mapping of maize stripe disease resistance from the
Mascarene source. Theor. Appl. Genet. 111: 347-359. https://doi.org/10.1007/s00122-005-2027-3.
Di Renzo M.A., Bonamico N.C., Díaz D.G., Ibañez M.A., Faricelli M.E., Balzarini
M.G., Salerno J.C. (2004) Microsatellite markers linked to QTL for resistance
to Mal de Río Cuarto disease in Zea mays L. J. Agric. Sci. 142:
289-295. https://doi.org/10.1017/S0021859604004307.
Di Renzo M.A., Bonamico N.C., Díaz D.G., Salerno J.C., Ibañez M.A., Gesumaria
J.J. (2002) Inheritance of resistance to Mal de Río Cuarto (MRC) disease in Zea
mays (L.) J. Agric. Sci. 139: 47-53. https://DOI:10.1017/S0021859602002241.
Distéfano A.J., Conci L.R., Muñoz Hidalgo M., Guzmán F.A., Hopp H.E., del
Vas M. (2002) Sequence analysis of genome segments S4 and S8 of Mal de Río
Cuarto virus (MRCV): evidence that the virus should be a separate Fijivirus species.
Arch. Virol. 147: 1699-1709.
Gimenez Pecci M.P. (2012) Mal de Río Cuarto del maíz. En: Gimenez Pecci M.P.,
Laguna I.G., Lenardón S.L. (Eds.) Enfermedades
del maíz producidas por virus y mollicutes en Argentina, INTA, Buenos Aires,
pp. 41-56.
Guo J., Pradhan S., Shahi D., Khan J., Mcbreen J., Bai G.J., Murphy P., Babar
M.A. (2020) Increased prediction accuracy using combined genomic information
and physiological traits in a soft wheat panel evaluated in multi-environments.
Sci. Rep. 10: 7023. https://doi.org/10.1038/s41598-020-63919-3.
Gutierrez L., Cuesta Marcos A., Castro A.J., von Zitzewitz J., Schmitt M.,
Hayes P.M. (2011) Association mapping of malting quality quantitative trait
loci in winter barley: positive signals from small germplasm arrays. Plant
Genome 4: 256-272.
Habier D., Fernando R., Kizilkaya K., Garrick D. (2011) Extension of the
Bayesian alphabet for genomic selection. BMC Bioinformatics 12: 186.
Hao D., Cheng Y., Chen G., Lu H., Shi M., Zhang Z., Huang X., Mao Y., Xue
L. (2015) Identification of significant single nucleotide polymorphisms for
resistance to maize rough dwarf disease in elite maize (Zea mays L.)
inbred lines. Euphytica 203: 109-20.
Holland J.B., Nyquist W.E., Cervantes Martínez C.T. (2010) Estimating
and Interpreting Heritability for Plant Breeding: An Update. Plant Breed. Rev. https://doi.org/10.1002/9780470650202.ch2.
Lapierre H., Signoret P.A. (2004) Viruses and virus diseases of Poaceae
(Gramineae). INRA ED, Paris.
Li J., Ji L. (2005) Adjusting multiple testing in multilocus analyses
using the eigen values of a correlation matrix. Heredity 95: 221-227. https://doi.org/10.1038/sj.hdy.6800717.
Ornaghi J.A., Boito G., Sanchez G., March G., Beviacqua J.E. (1993) Studies
on the populations of Delphacodes kuscheli Fennah in different years and
agricultural areas. J. Plant Genet. Breed. 47: 277-282.
Parisseaux B., Bernardo R. (2004) In silico mapping of quantitative
trait loci in maize. Theor. Appl. Genet. 109:
508-514. https://doi.org/10.1007/s00122-004-1666-0.
Perez Rodriguez P., de los Campos G. (2014) Genome-Wide Regression and
Prediction with the BGLR Statistical Package. Genetics 198 (2): 483-495.
Price A.L., Patterson N.J., Plenge R.M., Weinblatt M.E., Shadick N.A., Reich
D. (2006) Principal components analysis corrects for stratification in
genome-wide association studies. Nat. Genet. 38: 904-909. https://doi.org/10.1038/ng1847.
Pritchard J.K., Stephens M., Rosenberg N.A., Donnelly P. (2000) Association
mapping in structured populations. Am. J. Hum. Genet. 67: 170-181.
https://doi.org/10.1086/302959.
R Core Team (2016) R: A language and environment for statistical
computing. R Foundation for Statistical Computing, Vienna, Austria, URL (http://www.R-project.org/).
Redinbaugh M.G., Lübberstedt T., Leng P., Xu M. (2018) The genetics and
genomics of virus resistance in maize. In: Bennetzen J., Flint Garcia S., Hirsch
C., Tuberosa R. (Eds.) The Maize Genome. Compendium of Plant Genomes. Springer,
Cham.
Remes Lenicov A.M.M., Tesón A., Dagoberto E., Huguet N. (1985) Hallazgo de uno de los vectores del Mal
de Río Cuarto en maíz. Gaceta Agronómica 5: 251-258.
Rossi E.A., Borghi M.L., Di Renzo M.A., Bonamico N.C. (2015) Quantitative
Trait loci (QTL) Identification for Resistance to Mal de Río Cuarto Virus
(MRCV) in Maize Based on Segregate Population. Open Agric. J. 9: 48- 55.
Rossi E.A., Ruiz M., Bonamico N.C., Balzarini M.G. (2020) Genome-wide
association study of resistance to Mal de Río Cuarto disease in maize. Agronomy
J. https://doi.org/10.1002/agj2.20448.
Rossi E.A., Ruiz M., Di Renzo M., Bonamico N.C. (2019a) Genotypic
diversity in 291 maize lines from CIMMYT and phenotypic characterization in
southern Cordoba, Argentina. J. Basic Appl. Genet. 30:
25-33.
Rossi E.A., Ruiz M., Rueda Calderón M.A., Bruno C.I., Bonamico N.C., Balzarini
M.G. (2019b) Meta-analysis of QTL studies for resistance to fungi and viruses
in maize. Crop Sci. 59: 125-139. https://doi.org/10.2135/cropsci2018.05.0330.
Seem R.C. (1984) Disease Incidence and Severity Relationships. Annu.
Rev. Phytopathol. 22: 133-150. https://doi.org/10.1146/annurev.py.22.090184.001025.
Shi L., Hao Z.F., Weng J.F., Xie C.X., Liu C.L., Zhang D., Zhang S. (2012)
Identification of a major quantitative trait locus for resistance to maize
rough dwarf virus in a Chinese maize inbred line X178 using a linkage map based
on 514 gene-derived single nucleotide polymorphisms. Mol. Breed. 30: 615-625.
https://doi.org/10.1007/s11032-011-9652-0.
Turner D.S. (2018) qqman: an R package for visualizing GWAS results
using Q-Q and manhattan plots. J. Open Source Softw. 3: 731. https://doi.org/10.21105/joss.00731.
Warburton M., Tang J., Windham G., Hawkins L., Murray S., Xu W., Boykin D.,
Perkins A., Williams W. (2015) Genome-Wide Association Mapping of Aspergillus
flavus and Aflatoxin Accumulation Resistance in Maize. Crop Sci. 55: 1-11.
West B., Welch K.B., Galecki A.T. (2007) Linear mixed models: A
practical guide using statistical software. Chapman & Hall, Boca Raton, FL.
Wu Y., San Vicente F., Huang K., Dhliwayo T., Costich D.E., Semagn K., Babu
R. (2016) Molecular characterization of CIMMYT maize inbred lines with
genotyping by sequencing SNPs. Theor. Appl. Genet. 129:
753-765. https://doi.org/10.1007/s00122-016-2664-8.
Yu J., Holland J.B., McMullen M.D.,
Buckler E.S. (2008) Genetic design and statistical power of nested association
mapping in maize. Genetics 178: 539-551. https://doi.org/10.1534/genetics.107.074245.
Yu J., Pressoir G., Briggs W.H., Bi I.V., Yamasaki M., Doebley J.F., Buckler
E.S. (2006) A unified mixed model method for association mapping that accounts
for multiple levels of relatedness. Nat. Genet. 38: 203-208.
https://doi.org/10.1038/ng1702.
Zhao K., Aranzana M.J., Kim S., Lister C., Shindo C., Tang C., Nordborg M.
(2007) An Arabidopsis example of association mapping in structured samples. PLoS
Genet. 3: 71-82. https://doi.org/10.1371/journal.pgen.0030004.