


Vol. XXXII Issue 1
Article 3
DOI:
10.35407/bag.2021.32.01.03
ARTÍCULOS ORIGINALES
Modelos multivariados en la búsqueda de
regiones genómicas para resistencia a mal de río cuarto y bacteriosis en maíz
Multi-trait models for
genomic regions associated with mal de río cuarto and bacterial disease in
maize
Ruiz M. 1,2*
Rossi E.A.1,2
Bonamico N.C. 1,2
Balzarini M.G.3,4
1 INIAB-Instituto de Investigaciones Agrobiotecnológicas (CONICET-UNRC).
2 Facultad de Agronomía y Veterinaria, Universidad Nacional
de Río Cuarto.
3 CONICET-Consejo Nacional de Investigaciones Científicas y
Técnicas.
4 Facultad de Ciencias Agropecuarias, Universidad Nacional de
Córdoba.
* Corresponding
author: Marcos Ruiz mruiz@ayv.unrc.edu.ar
ORCID 0000-0001-5624-0183
RESUMEN
La producción de maíz (Zea Mays L.)
ha sido ampliamente beneficiada con la mejora de líneas endocriadas respecto a
la resistencia a enfermedades causadas por virus y hongos. Sin embargo, es
notable la ausencia de genotipos resistentes a bacteriosis. El objetivo del
presente estudio fue identificar regiones genómicas para la mejora de
resistencia a Mal de Río Cuarto (MRC) y a bacteriosis (BD) en un germoplasma
diverso de maíz. Se evaluó, para ambas enfermedades, una población diversa de
líneas de maíz en el ciclo de cultivo 2019-2020 en la región argentina donde la
virosis MRC es endémica. Se estimó incidencia y severidad de MRC y BD en cada
línea y se realizó un estudio de mapeo por asociación (GWAS) con 78.376
marcadores SNPs. Un modelo multicarácter se utilizó para evaluar
simultáneamente la resistencia a MRC y BD en las líneas evaluadas. El
germoplasma evidenció alta variabilidad genética tanto para la mejora de la
resistencia a MRC como a BD, pero no se observó correlación genética
significativa entre la respuesta a ambas enfermedades. Se identificaron
regiones genómicas promisorias para resistencia a MRC y a BD, que serán
confirmadas en evaluaciones en nuevos ambientes.
Palabras clave: Enfermedad en maíz; Mapeo
por asociación; SNP;
Modelo multivariado
ABSTRACT
Maize (Zea Mays L.) production has been greatly benefited from
the improvement of inbred lines in regard to the resistance to diseases.
However, the absence of resistant genotypes to bacteriosis is remarkable. The
aim of the study was to identify genomic regions for resistance to Mal de Río
Cuarto (MRC) and to bacterial disease (BD) in a diverse maize germplasm evaluated
in the Argentinian region where MRC virus is endemic. A maize diverse
population was assessed for both diseases during the 2019-2020 crop season.
Incidence and severity of MRC and BD were estimated for each line and a genome
wide association study (GWAS) was conducted with 78,376 SNP markers. A
multi-trait mixed linear model was used for simultaneous evaluation of
resistance to MRC and BD in the scored lines. The germplasm showed high genetic
variability for both MRC and BD resistance. No significant genetic correlation
was observed between the response to both diseases. Promising genomic regions for
resistance to MRC and BD were identified and will be confirmed in further
trials.
Key words: Maize disease; Genome wide
association study; SNP; Multi-trait model
Received: 06/25/2020
Revised version received: 10/30/2020
Accepted: 11/10/2020
INTRODUCCIÓN
El maíz es un importante cultivo a nivel
mundial con una producción aproximada de 1.148 millones de toneladas. Argentina
es el cuarto productor con un total de 43,5 millones de toneladas cosechadas en
el ciclo agrícola 2018/19. Entre otros factores, su producción es afectada por
la presencia de enfermedades que pueden amenazar el cultivo y consecuentemente
la seguridad alimentaria y la sustentabilidad agrícola (Nelson et al., 2018). Las enfermedades más comunes en maíz son
causadas por virus y hongos (Agrios, 2005) aunque entre las patologías emergentes, se
encuentran las producidas por bacterias. Las enfermedades producidas por
bacterias patógenas en maíz han incrementado su prevalencia en Argentina (Plazas, 2018), debido posiblemente a la masiva adopción de
la siembra directa. Es notable la ausencia de genotipos resistentes a este tipo
de enfermedades (Gurr y Rushton, 2005). La base genética de la respuesta a la
infección por bacterias o bacteriosis (BD) ha sido significativamente menos
investigada que aquélla relacionada con enfermedades fúngicas y virales en maíz
(Rossi et
al., 2019). Entre
las enfermedades virales, el Mal de Río Cuarto (MRC) juega un rol prevalente
para el cultivo de maíz en Argentina ya que ha causado severas pérdidas de rendimiento
con diferentes valores de incidencia y severidad a través de los años (Giménez
Pecci et al., 2012). El agente causal es el Mal de Río Cuarto virus (MRCV),
el cual se clasifica como un miembro del género Fijivirus, familia Reoviridae
(King et
al., 2012) y se
transmite de manera persistente propagativa por insectos vectores, principalmente
por la chicharrita Delphacodes kuscheli Fennah (Ornaghi et al., 1993).
La resistencia genética es el método más eficiente y efectivo para el control
de MRC y por ello se han realizado numerosas evaluaciones de material genético
en la zona donde la enfermedad es endémica (Di Renzo et al., 2004; Bonamico et al., 2012; Rossi et al.,
2015). Numerosos QTL (Quantitative trait loci) para resistencia a MRC se
han identificado a través del fenotipado de poblaciones biparentales generadas localmente.
El mapeo por asociación (GWAS, genome wide association study) también es
usado para la identificación de variantes alélicas específicas que incrementan
la resistencia a enfermedades (Zila et al., 2014). Para GWAS, es esencial contar con
poblaciones compuestas por individuos genotípica y fenotípicamente diversos,
que presenten una alta densidad de polimorfismos en la secuencia de ADN (Yan et al., 2011). Germoplasmas diversos de especies alógamas
como el maíz demandan alta densidad de marcadores. Esto se debe a que el
desequilibrio de ligamiento puede disminuir entre dos sitios polimórficos a una
distancia corta (unos pocos miles pares de bases) a causa de la alta frecuencia
de recombinación (Remington et al., 2001). Las líneas de maíz del Centro Internacional
de Mejoramiento de Maíz y Trigo (CIMMYT), desarrolladas durante los últimos 25
años, se han convertido en la principal fuente pública de germoplasma diverso
de maíz (Chen et al., 2016). La genotipificación densa de este
germoplasma de alta diversidad genómica aporta información para la
implementación de GWAS para identificar loci de resistencia a
enfermedades presentes en la región maicera de Argentina. Si bien es común que
los mejoradores evalúen múltiples caracteres en sus esquemas de selección (Malosetti et al., 2008) en programas de mejoramiento genético, la
evaluación de resistencia a MRC no se ha realizado simultáneamente con BD. El enfoque
multicarácter incrementa el poder de detección de aquellos QTL que afectan a
más de un carácter simultáneamente (Knott y Haley, 2000). El ajuste de un modelo que permita caracterizar
genotipos considerando más de un carácter y sus potenciales correlaciones (Covarrubias Pazaran, 2016) representa una forma novedosa de abordar la
resistencia a enfermedades. El objetivo del presente estudio fue identificar
regiones genómicas de maíz promisorias para la mejora de la resistencia a la enfermedad
MRC y a BD en un germoplasma diverso de maíz.
MATERIALES Y MÉTODOS
Material vegetal y ensayo de campo
Se sembró una población diversa de 185 líneas de maíz del Centro
Internacional de Mejoramiento de Maíz y Trigo (CIMMYT) para determinar su
resistencia a Mal de Río Cuarto (MRC) y a bacteriosis (BD) durante el ciclo de
cultivo 2019-2020 en Río Cuarto, Córdoba, Argentina (64° 20′ W, 33°
8′ S). Se utilizó un diseño parcialmente repetido (p-rep) (Cullis et al., 2006). El diseño implicó el uso de tres
repeticiones en 50 líneas (de modo que p= 27%) y parcelas individuales en las
135 líneas restantes. Cada línea se sembró en una parcela que consistió de un
surco de 2,5 m de longitud y 0,52 m de ancho. Se sembró a doble densidad y se
ralearon plantas, tres semanas posteriores a la emergencia para obtener 10 plantas
por parcela. La infección de ambas enfermedades ocurrió de manera natural.
Líneas experimentales susceptibles se evaluaron conjuntamente en el ensayo para
verificar la ocurrencia de MRC. Ambas enfermedades se evaluaron mediante la observación
de síntomas en la etapa de floración (95- 100 días post siembra). La incidencia
de MRC y BD se estimó como la proporción de plantas que presentaron síntomas
sobre el total de plantas de cada parcela (INCMRC y INC-BD, respectivamente).
La severidad en ambas enfermedades (SEV-MRC y SEV-BD) se evaluó utilizando las
escalas propuestas por Ornaghi et al. (1999) y Schuelter et al. (2003) para
MRC y BD, respectivamente. Para SEV-MRC, cada planta se clasificó por el grado
de severidad: 0= sin síntomas; 1= presencia de enaciones; 2= presencia de
enaciones + acortamiento de entrenudos + láminas foliares atrofiadas en el
tercio superior; 3= máximo desarrollo de la enfermedad con enaciones +
acortamiento de entrenudos + láminas foliares atrofiadas en el tercio superior
+ espigas pequeñas, múltiples y sin granos. Para SEV-BD, las plantas de cada parcela
se evaluaron visualmente por la lesión foliar, siendo 1= sin lesiones; 2=
lesiones dispersas; 3= hasta el 50% de las hojas con lesiones, con lesiones
graves en el 25% de las hojas inferiores; 4= hasta el 75% de las hojas con
lesiones, con lesiones graves en el 50% de las hojas inferiores; 5= 100% de las
hojas con lesiones, con lesiones graves en el 75% de las hojas inferiores; 6=
planta muerta. Para el análisis estadístico se usó la severidad promedio por
parcela.
Datos genómicos
La caracterización genotípica realizada
por Wu et al.
(2016) con marcadores
moleculares del tipo SNP utilizada en este trabajo se encuentra disponible en http://data.cimmyt.org/dvn.
De un total de 362.008 SNPs se seleccionaron 78.376 marcadores distribuidos en
los 10 cromosomas de maíz. La selección de marcadores se basó en la calidad, y
en un primer paso, se eliminaron marcadores con una frecuencia alélica menor a
0,05 o con errores de secuenciación. Posteriormente, se eliminaron los
marcadores con una tasa de datos faltantes superior al 35%. La base de datos
utilizada en este trabajo está disponible en
https://github.com/PlantbreedingUNRC/GWAS-MRC-BD.
Análisis estadístico
Los datos fenotípicos se analizaron
utilizando un modelo lineal mixto (MLM) multicarácter (Maier et al., 2015). La base de datos utilizada en el modelo
consta de 185 líneas de maíz evaluadas mediante cuatro caracteres (INCMRC, SEV-MRC,
INC-BD y SEV-BD). Se usó un modelo lineal mixto multivariado para obtener el
predictor lineal del efecto aleatorio de genotipo, es decir un BLUP multivariado
de efectos de genotipo que considera los cuatro caracteres evaluados
simultáneamente. La observación yijk para el carácter k es
escrito como una función lineal del efecto del bloque desde donde es observada
y del efecto de genotipo,
![]()
para el carácter k donde yijk es
la observación fenotípica para el carácter k=INC-MRC, SEV-MRC, INC-BD,
SEV-BD, μ es la media general, βi es el efecto
aleatorio deli-ésimobloque βi ~ N (0, σ2 b), Gj es
el efecto aleatorio del j-ésimogenotipo y εijk es el componente de
error aleatorio independiente que se supone normal con media y varianza
constante. El MLM multivariado es un modelo para el vector de observaciones Y
donde se consideran las observaciones de los k caracteres y que
estima no sólo varianzas de los caracteres sino también covarianzas entre
ellos. En este trabajo se ajustó utilizando la función “mmer” del paquete
“sommer” (CovarrubiasPazaran, 2016), software R (R Core Team, 2016). Los
componentes de varianza obtenidos del MLM multivariado fueron usados para
estimar la heredabilidad del carácter k, tal como lo propusieron Hallauer y Miranda (1988).

donde es la varianza genotípica para el
carácter k-ésimo, es la varianza residual asociada a las observaciones de ese
carácter, y p es una media ponderada del número de repeticiones (Holland et al., 2003). Las varianzas genéticas estimadas para cada carácter
y las covarianzas genética entre pares de caracteres, obtenidas del ajuste del
MLM multivariado, permitieron estimar la correlación genética (rg) entre los caracteres
l y m:

donde es la covarianza genética entre los
caracteres l y m, y los elementos del denominador corresponden a
las varianzas genéticas de ambos caracteres.
Asociación entre variación fenotípica y
variación genotípica
El software Tassel 5.2.60 (Bradbury et al., 2007) se utilizó para realizar el análisis de
asociación entre variación fenotípica y variación genotípica. El GWAS se llevó
a cabo con 78.376 SNPs. La incidencia y la severidad de MRC y BD fueron usadas
como variables dependientes. Para llevar a cabo la asociación se ajustaron seis
modelos. Los modelos fueron: 1) el modelo Naive, que realiza la
asociación de la información genotípica y fenotípica sin tener en cuenta la
posible existencia de estructura genética en la población en estudio; 2) el modelo
Q, que utiliza la matriz de estructura poblacional definida con el programa
Structure (Pritchard, 2000) considerando la correlación genética entre líneas; 3) el modelo
PCA, el cual modela la estructura poblacional subyacente en la población de
líneas de maíz, mediante la incorporación de cinco componentes principales (Price et al., 2006). También se ajustaron modelos que
incorporaron la matriz de Kinship para modelar la relación genética
entre dos líneas de la población en estudio, 4) el modelo K (Parisseaux y Bernardo, 2004); 5) el modelo Q + K (Yu et al., 2006); y 6) el modelo PCA + K (Zhao et al., 2007). A partir del gráfico de quantilesquantiles (Q-Q
plot) se seleccionaron los modelos apropiados para cada carácter. Este
gráfico compara los valores observados del -log10 (valor-p) para cada marcador
con los valores esperados del -log10 (valor-p) bajo la hipótesis nula de
no asociación entre marcadores moleculares y los caracteres en estudio. El
procedimiento de Li y Ji (2005) se utilizó para realizar correcciones de los
valores p por multiplicidad. Los marcadores SNPs que superaron el umbral
de -log10 (valor-p) >4 (p<0,0001), se consideraron
asociados de manera significativa con el carácter. Los gráficos Q-Q plot y
Manhattan plot se realizaron con el paquete “qqman” (Turner, 2018) del software R (R Core Team, 2016) utilizando
las salidas de GWAS del software Tassel.
RESULTADOS
Para ambas enfermedades se observaron
tanto valores mínimos como valores máximos de las respectivas escalas de
evaluación de síntomas. El valor medio de INCMRC fue 43% y de SEV-MRC 0,88,
mientras que para INC-BD y SEV-BD fueron 83% y 2,5, respectivamente. El valor
estimado de heredabilidad varió entre 0,35 (INC-BD) y 0,7 (SEV-BD) (Tabla 1).
Tabla 1. Medidas resumen y parámetros genéticos
estimados para incidencia (INC) y severidad (SEV) de Mal de Río Cuarto (MRC) y
de bacteriosis (BD) en una población diversa de 185 líneas de maíz evaluadas en
Río Cuarto, Córdoba, Argentina, durante el ciclo de cultivo 2019/2020.

El MLM multivariado reveló que la
correlación genética entre caracteres es positiva y significativa cuando se
trata de INC y SEV de una misma enfermedad, mientras que la correlación entre
caracteres de distinta enfermedad no resultó estadísticamente significativa (Tabla
2).
Tabla 2. Correlación entre los caracteres incidencia
(INC) y severidad (SEV) de Mal de Río Cuarto (MRC) y de bacteriosis (BD) en una
población diversa de 185 líneas de maíz evaluadas en Río Cuarto, Córdoba,
Argentina, en el ciclo de cultivo 2019/2020.

*correlación significativa (p < 0,0001)
INC-MRC: Incidencia de Mal de Río Cuarto, SEV-MRC: Severidad de Mal de Río
Cuarto, INC-BD: Incidencia de bacteriosis, SEV-BD: severidad de bacteriosis.
A partir de la frecuencia alélica de los
loci de cada línea, se clasificaron con el software Structure los genotipos del
panel en tres grupos. El primer grupo estuvo compuesto por cuatro líneas, el
segundo por 108 líneas y el tercer grupo por 73 líneas. Consecuentemente, la
estructura genética que determina el agrupamiento de líneas fue contemplada en
los modelos de asociación entre la variación fenotípica y la genotípica
ajustadas carácter por carácter. El modelo Q (modelo que considera la
estructura de grupos sugerida por el software Structure) fue el modelo de mejor
ajuste para INCMRC y SEV-MRC. Sin embargo, para INC-BD y SEVBD la estructura de
grupos fue menos discreta y quedó mejor representada por el modelo de
asociación PCA + K, modelo que consideró cinco componentes del análisis de
componentes principales de los datos moleculares como covariable y que además
incorpora la correlación genética entre pares de líneas mediante la matriz de parentesco
K (Figura 1). El mapeo asociativo utilizando los 78.376
SNPs para GWAS, se realizó con el modelo más apropiado para cada carácter (Figura 1).

Figura 1. Gráficos Q-Q plot y Manhattan plot para
el GWAS de los caracteres incidencia y severidad de Mal de Río Cuarto y de
bacteriosis en una población diversa de 185 líneas de maíz. a) INC-MRC, b)
SEV-MRC, c) INC-BD, d) SEV-BD. Umbral -log10 (0,0001).
Cinco regiones fueron declaradas como
estadísticamente significativas tanto en el carácter INC-MRC como para SEV-MRC.
Se identificaron otras 15 regiones como aportantes de alelos promisorios para
INC-BD y SEV-BD. No se encontraron regiones genómicas que fuesen promisorias
para mejorar la resistencia de ambas enfermedades simultáneamente. La
proporción de variación fenotípica explicada por cada región cromosómica
identificada como de alto potencial para la selección por resistencia a MRC
osciló entre 0,11 y 0,19; valores similares fueron observados para BD (Tabla 3
y 4).
Tabla 3. Marcadores moleculares asociados con incidencia
(INC) y severidad (SEV) de Mal de Río Cuarto en una población diversa de 185
líneas de maíz evaluadas en Río Cuarto, Córdoba, Argentina, en el ciclo de
cultivo 2019/2020.
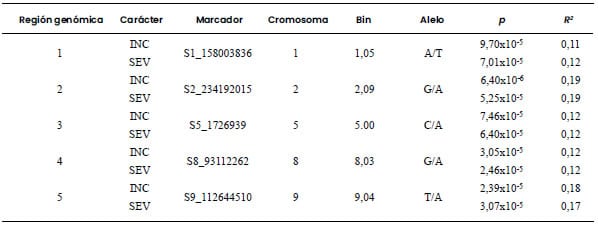
Umbral de significancia: valor-p < 0,0001.
Tabla 4. Marcadores moleculares asociados con incidencia
(INC) y severidad (SEV) de bacteriosis en una población diversa de 185 líneas
de maíz evaluadas en Río Cuarto, Córdoba, Argentina, en el ciclo de cultivo
2019/2020.
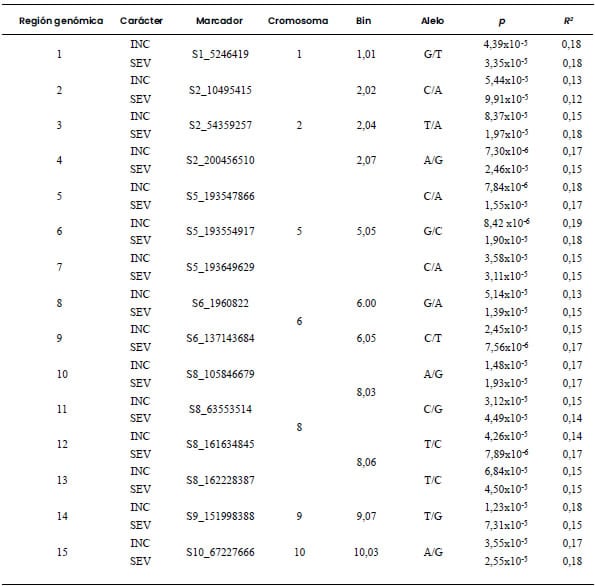
Umbral de significancia: valor-p < 0,0001.
DISCUSIÓN
La población diversa de líneas de maíz
del CIMMYT evaluada en Río Cuarto, presentó amplia variabilidad fenotípica y
genotípica respecto a la reacción de las líneas para resistencia a Mal de Río
Cuarto (MRC) y a bacteriosis (BD). Los modelos mixtos ofrecen un enfoque apropiado
para analizar conjuntamente los caracteres evaluados para la identificación de
mejores genotipos considerando la posible correlación entre éstos (Malosetti et al., 2008). La correlación entre caracteres puede ser
positiva, negativa o nula. Wisser et al. (2011) y López Zuniga et al. (2019)
observaron correlación positiva en caracteres de resistencia medido para tres enfermedades
causadas por Cochliobolus heterostrophus, Setosphaeria turcica, y
Cercosporazeae maydis en maíz. Sin embargo, en nuestro estudio no se
observó correlación entre la respuesta al MRC y a la BD. Consecuentemente, el
MLM multivariado constituye una herramienta útil para evaluar simultáneamente
INC y SEV de MRC o de BD, pero no para el tratamiento de ambas enfermedades ya
que la correlación genética entre resistencia a MRC y a BD fue baja. Los
síntomas de BD se desarrollan principalmente en las hojas, mientras que, los
síntomas de MRC afectan toda la planta cuando el ataque es severo (Abdala et al., 1997). La reducción de la lámina foliar puede,
consecuentemente, dificultar la evaluación de la resistencia a bacteriosis
produciendo subestimaciones en las correlaciones genéticas entre caracteres. Respecto
a la estructura genética poblacional detectada por Structure, observamos alta
consistencia con el agrupamiento de líneas como Lowland, Subtropical y Highland
propuesto por Wu et al. (2016), quienes trabajaron con un set de líneas en
el cual están contenidas las líneas del presente estudio. Al igual que lo
observado por Gutiérrez et al. (2015), no hubo un único modelo GWAS que ajustara
adecuadamente para todos los caracteres.
El modelo que mejor ajustó para los caracteres INC-MRC y SEV-MRC fue el modelo
Q (Pritchard,
2000); mientras que para INC-BD y SEV-BD ajustó
el modelo PCA + K (Zhao et al., 2007) que sugiere un agrupamiento más difuso y más
dependiente de las relaciones de pares de líneas que el modelo Q. El análisis
de GWAS permitió identificar regiones genómicas previamente reportadas para la
presencia de genes de resistencia a enfermedades. Di Renzo et
al. (2004) y Bonamico et al. (2012) detectaron
una región genómica que confiere resistencia a MRC en el bin 8.03 en una
población segregante F2:3 y en una población de RIL, ambas derivadas del
cruzamiento entre las líneas de maíz Mo17 (susceptible) y BLS14 (resistente). Gowda et al. (2015), detectó una región genómica en el bin 1.05 para
resistencia a Maize lethal necrosis disease en un germoplasma tropical
de maíz. Gomes de Paula Lana et al. (2017), evaluaron la resistencia a bacteriosis, Maize
white spot, en una población biparental y al igual que en el presente estudio
detectaron regiones genómicas asociadas para resistencia en los bin 2.07 y
8.03. Rossi et
al. (2020)
informaron dos regiones genómicas asociadas para resistencia a Maize white
spot en los bin1.01 y 8.03 a partir de una población de líneas tropicales
de maíz. Coincidentemente, estas dos regiones fueron asociadas significativamente
con resistencia a bacteriosis en el presente estudio donde se utilizó una
población diversa de diferente procedencia.
La variabilidad fenotípica y genotípica
existente en el germoplasma diverso constituido por las líneas de maíz de
CIMMYT resulta importante para apoyar programas de mejoramiento genético de
maíz locales. El GWAS detectó regiones genómicas con alelos promisorios tanto para
MRC como para BD, que individualmente explican una proporción de la variación
fenotípica que oscila entre 10 y 20% de la variación total, pero no se
identificaron regiones genómicas comunes a ambas enfermedades. Estudios
multiambientales permitirán confirmar si estas regiones genómicas resultan
promisorias para incorporar resistencia a Mal de Río Cuarto y a bacteriosis en
programas locales de mejoramiento genético de maíz.
BIBLIOGRAFÍA
Abdala G., Vigliocco A., Boito G., Lorenzo E. (1997) Dwarfism in Mal de
Río Cuarto disease. Histology of maize stems and endogenous gibberellin levels.
Biocell. 20: 211-220.
Agrios G. (2005) Plant pathology. Academic Press.
Bonamico N.C., Di Renzo M.A., Ibañez M.A., Borghi M.L., Díaz D.G., Salerno
J.C., Balzarini M.G. (2012) QTL analysis of resistance to Mal de Río Cuarto disease
in maize using recombinant inbred lines. J. Agric. Sci. 150 (5): 619-629.
https://doi.org/10.1017/ S0021859611000943.
Bradbury P., Zhang Z., Kroon D., Casstevens T., Ramdoss Y., Buckler E. (2007)
Tassel: Software for association mapping of complex traits in diverse samples. Bioinformatics
23: 2633- 2635.
Chen J., Zavala C., Ortega N., Petroli C., Franco J., Burgueño J., Costich
D., Hearne S.J. (2016) The development of quality control genotyping
approaches: A case study using elite maize lines. PLoS ONE 11 (6): 1-17.
https://doi.org/10.1371/journal.pone.0157236.
Covarrubias Pazaran G. (2016) Genome assisted prediction of quantitative
traits using the R package sommer. PLoS ONE 11 (6): e0156744.
https://doi:10.1371/journal.pone.0156744.
Cullis B.R., Smith A.B., Coombes N.E. (2006) On the design of early generation
variety trials with correlated data . J. Agric. Biol.
Environ. Stat. 11 (4): 381-393. https://doi.org/10.1198/108571106X154443.
Di Renzo M.A., Bonamico N.C., Díaz D.G., Ibañez M.A., Faricelli M.E., Balzarini
M.G., Salerno J.C. (2004) Microsatellite markers linked to QTL for resistance
to Mal de Río Cuarto disease in Zea mays L. J. Agric. Sci. 142
(3): 289-295. https://doi.org/10.1017/S0021859604004307.
Gimenez Pecci M. P. (2012) Mal de Río Cuarto del maíz. In: Gimenez Pecci
M.P., Laguna I.G., Lenardón S.L. (Eds.) Enfermedades
del maíz producidas por virus y mollicutes en Argentina. INTA, Buenos Aires, pp.
41-56.
Gomes de Paula Lana U., Prazeres de Souza I.R., Noda R.W., Pastina M.M.,
Vieira Magalhaes J., Teixeira Guimaraes C. (2017) Quantitative trait loci and resistance
gene analogs associated with maize white spot resistance. Plant Dis. 101: 200-208.
doi:10.1094/PDIS- 06-16-0899-RE.
Gowda M., Das B., Makumbi D., Babu R., Semagn K., Mahuku G., Olsen M.S.,
Bright J.M., Beyene Y., Prasanna B.M. (2015) Genome wide association and genomic
prediction of resistance to maize lethal necrosis disease in tropical maize
germplasm. Theor. Appl. Genet. 128 (10): 1957-1968. https://doi.org/10.1007/s00122-015-2559-0.
Gurr S.J., Rushton P.J. (2005) Engineering plants with increased disease
resistance: what are we going to express? Trends Biotechnol. 23: 275- 282.
Gutiérrez L., Germán S., Pereyra S., Hayes P.M., Pérez C.A., Capettini F.,
Locatelli A., Berberian H.M., Falconi E.E., Estrada R., Fros D., Gonza V., Altamirano
H., Huerta Espino J., Neyra E., Orjeda G., Sandoval Islas S., Sing R., Turkington
K., Castro A.J. (2015) Multienvironment multi-QTL association mapping
identifies disease resistance QTL in barley germplasm from Latin America. Theor. Appl. Genet. 128: 501- 519.
Hallauer A.R., Miranda J.B. (1988) Quantitative genetics in maize breeding,
2nd Edn. Iowa State University Press, Ames, IA.
Holland J.B., Nyquist W.E., Cervantes Martinez C.T. (2003) Estimating and
interpreting heritability for plant breeding: an update. Plant Breed. Rev. 22: 9-111.
King A.M.Q., Adams M.J., Carstens E.B., Lefkowitz E.J. (2012) Virus
taxonomy: classification and nomenclature of viruses: Ninth Report of the International
Committee on Taxonomy of Viruses. Elsevier Academic Press.
Knott S.A., Haley C.S. (2000) Multitrait least squares for quantitative
trait loci detection. Genetics 156 (2): 899-911.
Li J., Ji L. (2005) Adjusting multiple testing in multilocus analyses
using the eigen values of a correlation matrix. Heredity 95: 221-227.
https://doi.org/10.1038/sj.hdy.6800717.
Lopez Zuniga L.O., Wolters P., Davis S., Weldekidan T., Kolkman J.M., Rebecca
N., Hooda K.S., Rucker E., Thomason W., Wisser R., Balint Kurti P. (2019) Using
maize chromosome segment substitution line populations for the identification
of loci associated with multiple disease resistance. G3: Genes, Genom. Genet. 9
(1): 189-201. https://doi.org/10.1534/g3.118.200866.
Maier R., Moser G., Chen G.B., Ripke S., Coryell W., Potash J.B., Scheftner
W.A., Shi J., Weissman M.M., Hultman C.M., Landén M., Levinson D.F., Kendler K.S.,
Smoller J.W., Wray N.R., Lee S.H. (2015) Joint Analysis of Psychiatric Disorders
Increases Accuracy of Risk Prediction for Schizophrenia, Bipolar Disorder, and
Major Depressive Disorder. Am. J. Hum. Genet. 96: 283-294.
Malosetti M., Ribaut J.M., Vargas M., Crossa J., Van Eeuwijk F.A. (2008)
A Multi-trait multi-environment QTL mixed model with an application to drought
and nitrogen stress trials in maize (Zea Mays L.). Euphytica 161 (1- 2):
241-57. https://doi.org/10.1007/s10681-007-9594-0.
Nelson R., Wiesner Hanks T., Wisser R., Balint Kurti P. (2018) Navigating
complexity to breed disease-resistant crops. Nat. Rev. Genet. 19: 21-33. doi:10.1038/nrg.2017.82.
Ornaghi J.A., Boito G., Sanchez G., March G., Beviacqua J.E. (1993) Studies
on the populations of Delphacodes kuscheli Fennah in different years and
agricultural areas. J. Plant Genet. Breed. 47: 277-282.
Ornaghi J.A., March G.J., Boito G.T., Marinelli A., Beviacqua J.E., Giuggia
J., Lenardon S.L. (1999) Infectivity in natural populations of Delphacodes kuscheli
vector of “Mal Río Cuarto” Virus. Maydica. 44: 219-223.
Parisseaux B., Bernardo R. (2004) In silico mapping of quantitative
trait loci in maize. Theor. Appl. Genet. 109:
508- 514.
Plazas M.C., De Rossi R.L., Brücher E., Guerra F.A., Vilaró M., Guerra G.D.,
Wu G., Ortiz Castro M.C., Broders K. (2018) First report of Xanthomonas
vasicola pv. vasculorum causing bacteria leaf streak of maize (Zea Mays)
in Argentina. Plant Dis. 102 (5): 1026. https://doi.org/10.1094/PDIS-10-17-1578-PDN.
Price A., Patterson N., Plenge R., Weinblatt M., Shadick N., Reich D. (2006)
Principal components analysis corrects for stratification in genomewide association
studies. Nat. Genet. 38: 904-909.
Pritchard J., Stephens M., Rosenberg N., Donnelly P. (2000) Association mapping
in structured populations. Am. J. Hum. Genet. 67: 170-181.
R Core Team (2016) R: a language and environment for statistical
computing. R Foundation for Statistical Computing, Vienna, Austria.
Remington D.L., Thornsberry J.M., Matsuoka Y., Wilson L.M., Whitt S.R., Doebley
J., Kresovich S., Goodman M.M., Buckler E.S. (2001) Structure of linkage
disequilibrium and phenotypic associations in the maize genome. Proc. Natl.
Acad. Sci. U.S.A. 98 (20): 11479-84. https://doi.org/10.1073/pnas.201394398.
Rossi E.A., Borghi M.L., Di Renzo M.A., Bonamico N.C. (2015) Quantitative
trait loci (QTL) identification for resistance to Mal de Río Cuarto
virus (MRCV) in maize based on segregate population. Open Agric. J. 9: 48-55.
doi:10.2174/1874331501509010048.
Rossi E.A., Ruiz M., Rueda Calderón M.A., Bruno C.I., Bonamico N.C., Balzarini
M.G. (2019) Metaanalysis of loci for resistance to maize diseases. Crop Sci. 59:
1-15.
Rossi E.S., Kuki M.C., Pinto R.J.B., Scapim C.A., Faria M.V., De Leon N.
(2020) Genomic-wide association study for white spot resistance in a tropical maize
germplasm. Euphytica 216 (1). https://doi.org/10.1007/s10681-019-
2550-y.
Schuelter A.R., Prazeres De Souza I.R., Tavares F., Dos Santos M.X., Oliveira
E., Guimarães C.T. (2003) Phaeosphaeria genetic control of maize
resistance to Phaeosphaeria. Rev. Bras. Milho e Sorgo 2 (1): 80-86.
Turner S.D. (2018) qqman: an R package for visualizing GWAS results
using Q-Q and manhattan plots. J. Open Source Softw.
Wisser R.J., Kolkman J.M., Patzoldt M.E., Holland J.B., Yu J., Krakowsky
M., Nelson R.J., Balint Kurti P.J. (2011) Multivariate analysis of maize
disease resistances suggests a pleiotropic genetic basis and implicates a GST gene. Proc. Natl. Acad. Sci. U.S.A. 108 (18): 7339-44.
https://doi.org/10.1073/pnas.1011739108.
Wu Y., San Vicente F., Huang K., Dhliwayo T., Costich D.E., Semagn K., Sudha
N., Olsen M., Prassana B.M., Zhang X., Babu R. (2016) Molecular
characterization of CIMMYT maize inbred lines with genotyping by sequencing
SNPs. Theor. Appl. Genet. 129 (4): 753-65. https://doi.org/10.1007/s00122-016-
2664-8.
Yan J.B., Warburton M., Crouch J. (2011) Association mapping for enhancing
maize (Zea mays L.) genetic improvement. Crop Sci. 51: 433-49.
https://doi: 10.2135/cropsci2010.04.0233.
Yu J., Pressoir G., Briggs W., Bi I., Yamasaki M., Doebley J., McMullen M.,
Gaut B., Holland J., Kresovich S., Buckler E. (2006) A unified mixed-model
method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38: 203-208.
Zhao K., Aranzana M., Kim S., Lister C., Shindo C., Tang C., Toomajian C.,
Zheng H., Dean C., Marjoram P., Nordborg M. (2007) An Arabidopsis example
of association mapping in structured samples. PLoS Genet. 3: e4.
Zila C.T., Ogut F., Romay M.C., Gardner C.A., Buckler E.S., Holland J.B.
(2014) Genome-wide association study of fusarium ear rot disease in the U.S.A. Maize
inbred line collection. BMC Plant Biol.14 (1): 1-15. https://doi.org/10.1186/s12870-014-0372-6.